Das Saurav, Bora Sudipta Sankar, Yadav R N S, Barooah Madhumita
Department of Agricultural Biotechnology, Assam Agricultural University, Jorhat, Assam, India.
Centre for Studies in Biotechnology, Dibrugarh University, Dibrugarh, Assam, India.
Genom Data. 2017 Mar 30;12:89-96. doi: 10.1016/j.gdata.2017.03.013. eCollection 2017 Jun.
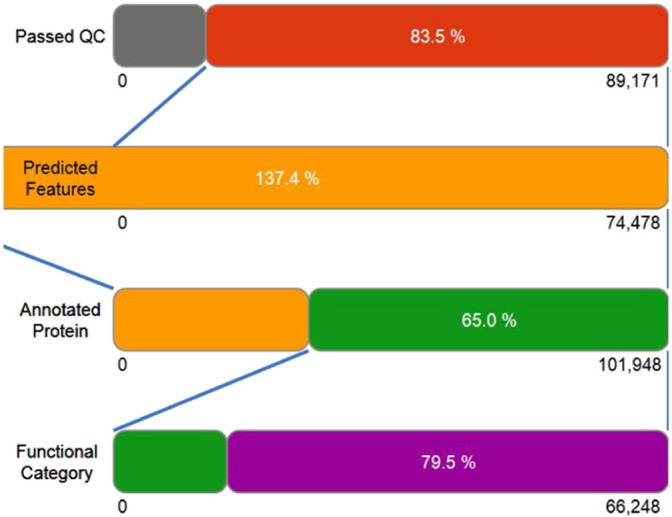
Metagenomic approach was used to understand the structural and functional diversity present in arsenic contaminated groundwater of the Ganges Brahmaputra Delta aquifer system. A metagene dataset (coded as TTGW1) of 89,171 sequences (totaling 125,449,864 base pairs) with an average length of 1406 bps was annotated. About 74,478 sequences containing 101,948 predicted protein coding regions passed the quality control. Taxonomical classification revealed abundance of bacteria that accounted for 98.3% of the microbial population of the metagenome. Eukaryota had an abundance of 1.1% followed by archea that showed 0.4% abundance. In phylum based classification, Proteobacteria was dominant (62.6%) followed by Bacteroidetes (11.7%), Planctomycetes (7.7%), Verrucomicrobia (5.6%), Actinobacteria (3.7%) and Firmicutes (1.9%). The Clusters of Orthologous Groups (COGs) analysis indicated that the protein regulating the metabolic functions constituted a high percentage (18,199 reads; 39.3%) of the whole metagenome followed by the proteins regulating the cellular processes (22.3%). About 0.07% sequences of the whole metagenome were related to genes coding for arsenic resistant mechanisms. Nearly 50% sequences of these coded for the arsenate reductase enzyme (EC. 1.20.4.1), the dominant enzyme of operon. Proteins associated with iron acquisition and metabolism were coded by 2% of the metagenome as revealed through SEED analysis. Our study reveals the microbial diversity and provides an insight into the functional aspect of the genes that might play crucial role in arsenic geocycle in contaminated ground water of Assam.
采用宏基因组学方法来了解恒河-布拉马普特拉河三角洲含水层系统受砷污染地下水中存在的结构和功能多样性。对一个包含89171条序列(总计125449864个碱基对)、平均长度为1406个碱基对的宏基因数据集(编码为TTGW1)进行了注释。约74478条包含101948个预测蛋白质编码区的序列通过了质量控制。分类学分析表明,细菌数量众多,占宏基因组微生物种群的98.3%。真核生物占比1.1%,古菌占比0.4%。在基于门的分类中,变形菌门占主导地位(62.6%),其次是拟杆菌门(11.7%)、浮霉菌门(7.�%)、疣微菌门(5.6%)、放线菌门(3.7%)和厚壁菌门(1.9%)。直系同源基因簇(COG)分析表明,调节代谢功能的蛋白质在整个宏基因组中占比很高(18199条读数;39.3%),其次是调节细胞过程的蛋白质(22.3%)。整个宏基因组中约0.07%的序列与编码抗砷机制的基因相关。这些序列中近50%编码砷酸还原酶(EC. 1.20.4.1),这是操纵子的主要酶。通过SEED分析发现,与铁获取和代谢相关的蛋白质由宏基因组的2%编码。我们的研究揭示了微生物多样性,并深入了解了在阿萨姆邦受污染地下水中砷地球化学循环中可能起关键作用的基因的功能方面。