Coffey Sydney R, Bragg Robert M, Minnig Shawn, Ament Seth A, Cantle Jeffrey P, Glickenhaus Anne, Shelnut Daniel, Carrillo José M, Shuttleworth Dominic D, Rodier Julie-Anne, Noguchi Kimihiro, Bennett C Frank, Price Nathan D, Kordasiewicz Holly B, Carroll Jeffrey B
Behavioral Neuroscience Program, Psychology Department, Western Washington University, Bellingham, WA, United States of America.
Institute for Genome Sciences and Department of Psychiatry, University of Maryland School of Medicine, Baltimore, MD, United States of America.
PLoS One. 2017 Apr 28;12(4):e0175968. doi: 10.1371/journal.pone.0175968. eCollection 2017.
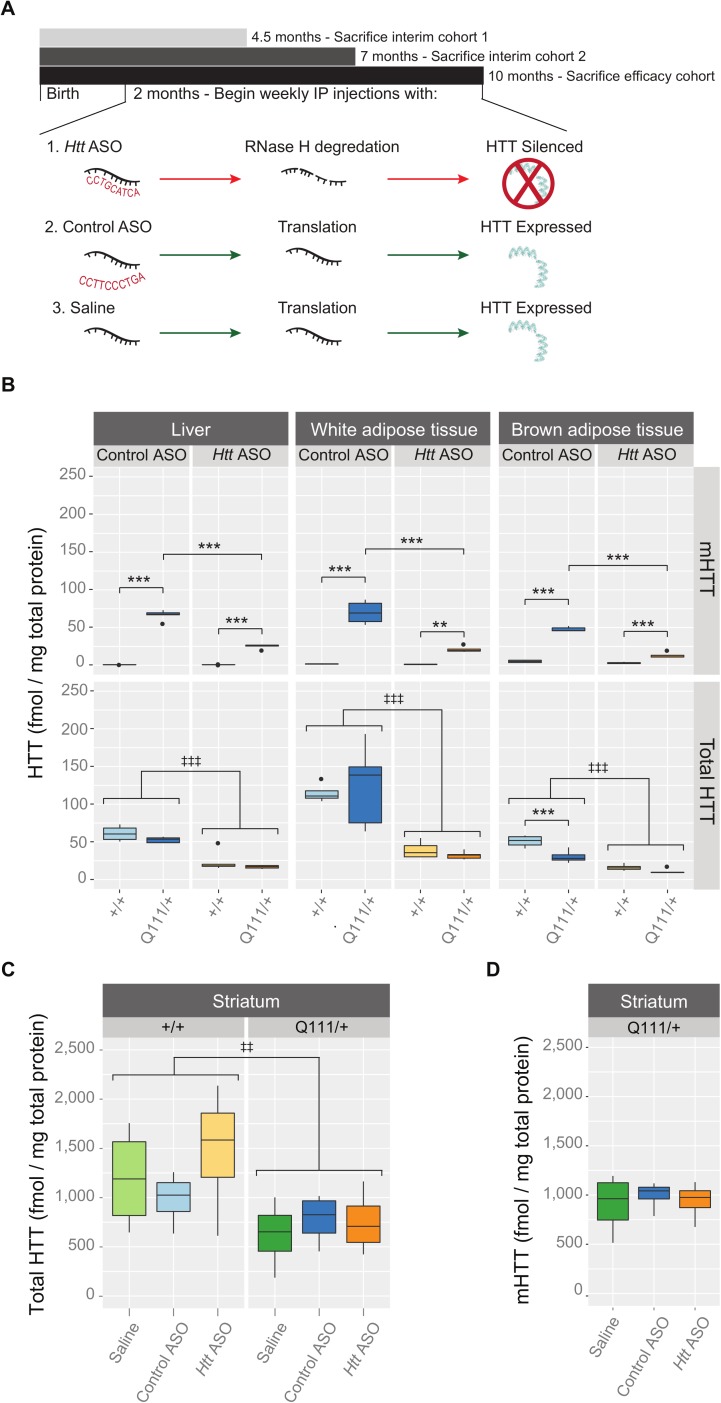
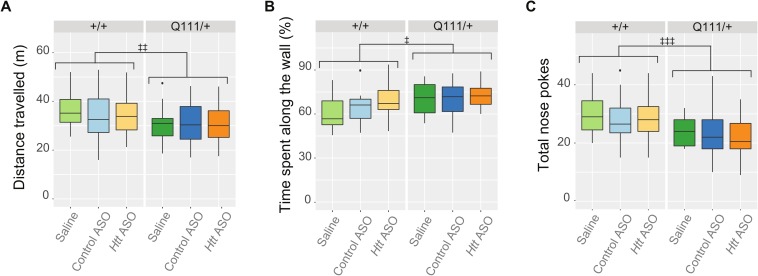
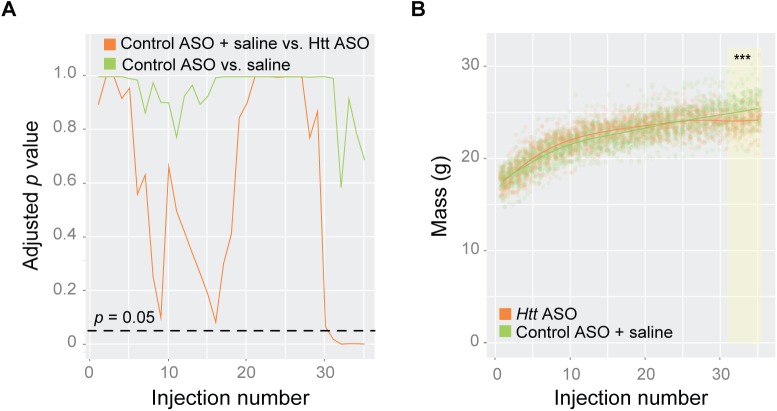
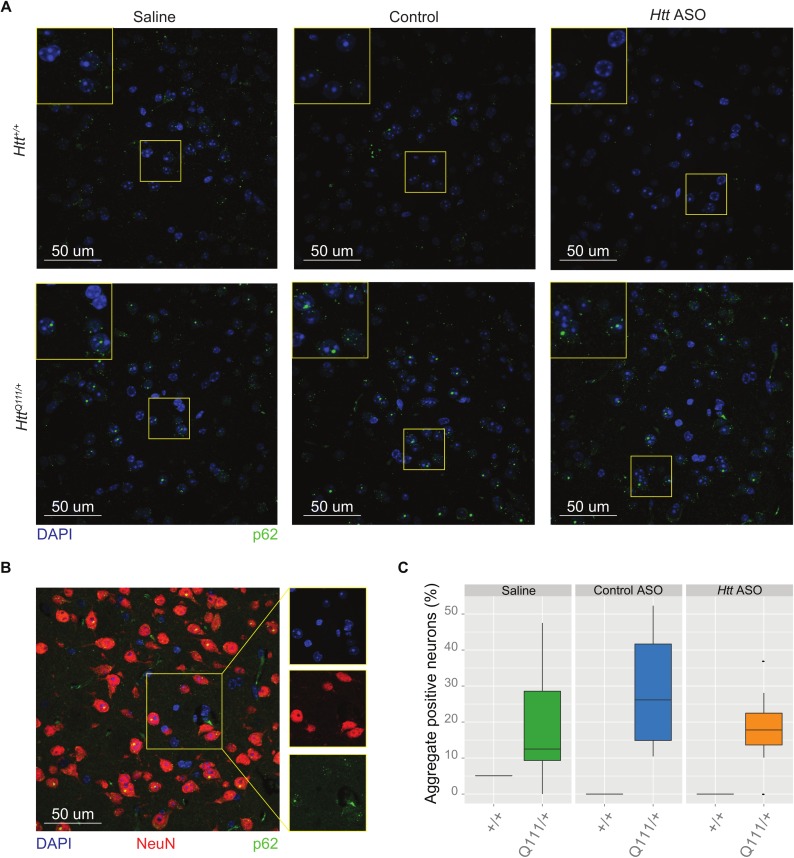
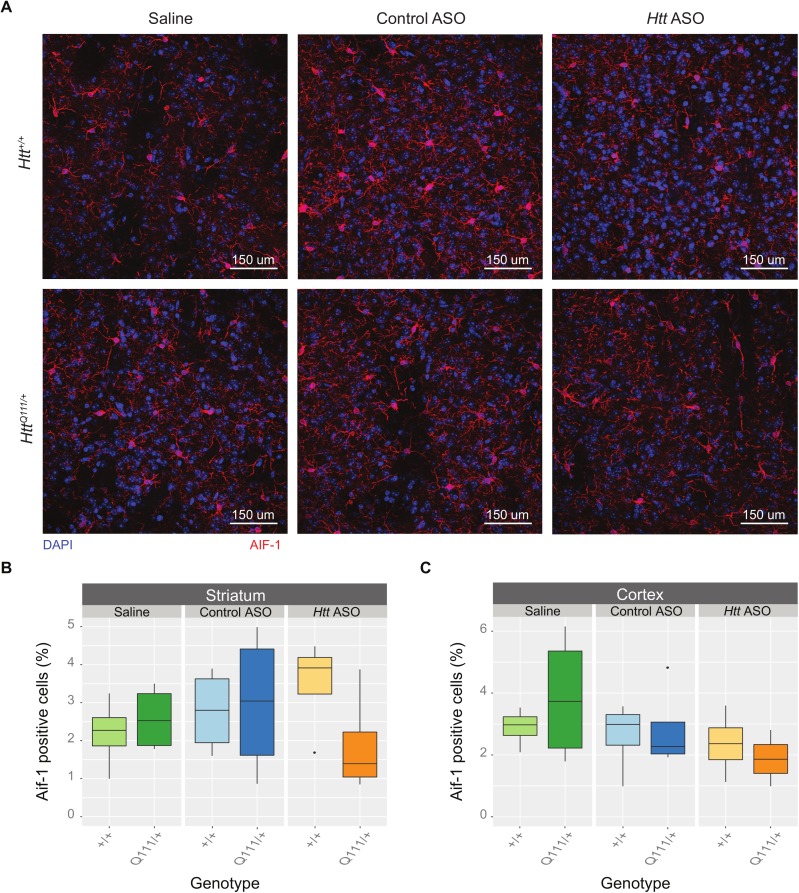
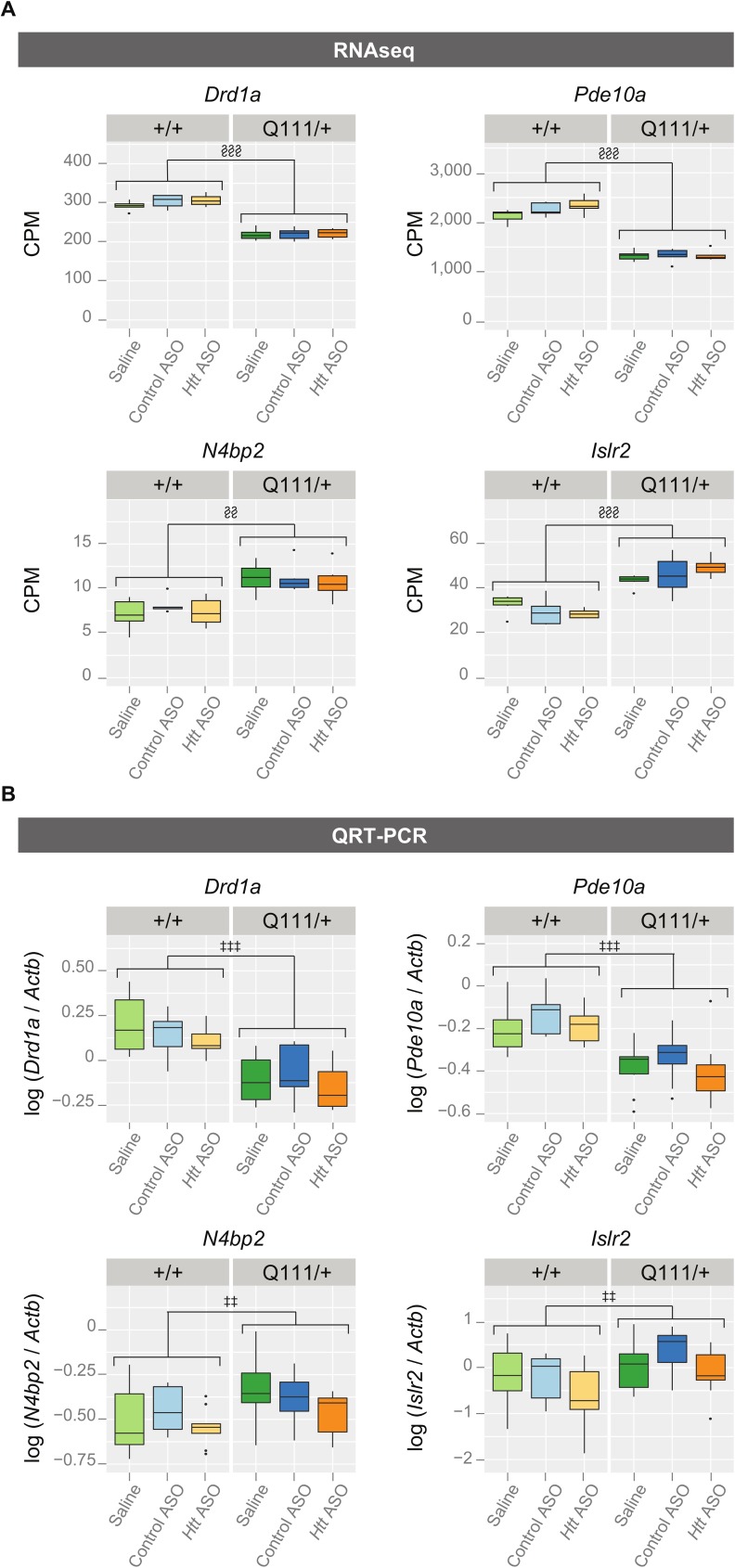
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease whose predominant neuropathological signature is the selective loss of medium spiny neurons in the striatum. Despite this selective neuropathology, the mutant protein (huntingtin) is found in virtually every cell so far studied, and, consequently, phenotypes are observed in a wide range of organ systems both inside and outside the central nervous system. We, and others, have suggested that peripheral dysfunction could contribute to the rate of progression of striatal phenotypes of HD. To test this hypothesis, we lowered levels of huntingtin by treating mice with antisense oligonucleotides (ASOs) targeting the murine Huntingtin gene. To study the relationship between peripheral huntingtin levels and striatal HD phenotypes, we utilized a knock-in model of the human HD mutation (the B6.HttQ111/+ mouse). We treated mice with ASOs from 2-10 months of age, a time period over which significant HD-relevant signs progressively develop in the brains of HttQ111/+ mice. Peripheral treatment with ASOs led to persistent reduction of huntingtin protein in peripheral organs, including liver (64% knockdown), brown adipose (66% knockdown), and white adipose tissues (71% knockdown). This reduction was not associated with alterations in the severity of HD-relevant signs in the striatum of HttQ111/+ mice at the end of the study, including transcriptional dysregulation, the accumulation of neuronal intranuclear inclusions, and behavioral changes such as subtle hypoactivity and reduced exploratory drive. These results suggest that the amount of peripheral reduction achieved in the current study does not significantly impact the progression of HD-relevant signs in the central nervous system.
亨廷顿舞蹈症(HD)是一种常染色体显性神经退行性疾病,其主要神经病理学特征是纹状体中中等棘状神经元的选择性丧失。尽管存在这种选择性神经病理学现象,但迄今为止,在几乎所有已研究的细胞中都发现了突变蛋白(亨廷顿蛋白),因此,在中枢神经系统内外的广泛器官系统中都观察到了相关表型。我们和其他人都曾提出,外周功能障碍可能会导致HD纹状体表型的进展速度加快。为了验证这一假设,我们通过用靶向小鼠亨廷顿基因的反义寡核苷酸(ASO)处理小鼠来降低亨廷顿蛋白的水平。为了研究外周亨廷顿蛋白水平与纹状体HD表型之间的关系,我们利用了人类HD突变的基因敲入模型(B6.HttQ111/+小鼠)。我们在2至10月龄期间用ASO处理小鼠,在此期间,HttQ111/+小鼠大脑中会逐渐出现与HD相关的明显体征。外周给予ASO导致外周器官中亨廷顿蛋白持续减少,包括肝脏(敲低64%)、棕色脂肪(敲低66%)和白色脂肪组织(敲低71%)。在研究结束时,这种减少与HttQ111/+小鼠纹状体中与HD相关体征的严重程度改变无关,这些体征包括转录失调、神经元核内包涵体的积累以及行为变化,如轻微活动减少和探索驱动力降低。这些结果表明,在本研究中实现的外周减少量并未显著影响中枢神经系统中与HD相关体征的进展。