McCormack Shana E, Li Dong, Kim Yeon Joo, Lee Ji Young, Kim Soo-Hyun, Rapaport Robert, Levine Michael A
Division of Endocrinology and Diabetes, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104.
Center for Applied Genomics, Department of Pediatrics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104.
J Clin Endocrinol Metab. 2017 Jul 1;102(7):2501-2507. doi: 10.1210/jc.2017-00332.
Pituitary stalk interruption syndrome (PSIS, ORPHA95496) is a congenital defect of the pituitary gland characterized by the triad of a very thin/interrupted pituitary stalk, an ectopic (or absent) posterior pituitary gland, and hypoplasia or aplasia of the anterior pituitary gland. Complex genetic patterns of inheritance of this disorder are increasingly recognized.
The objective of this study was to identify a genetic cause of PSIS in an affected child.
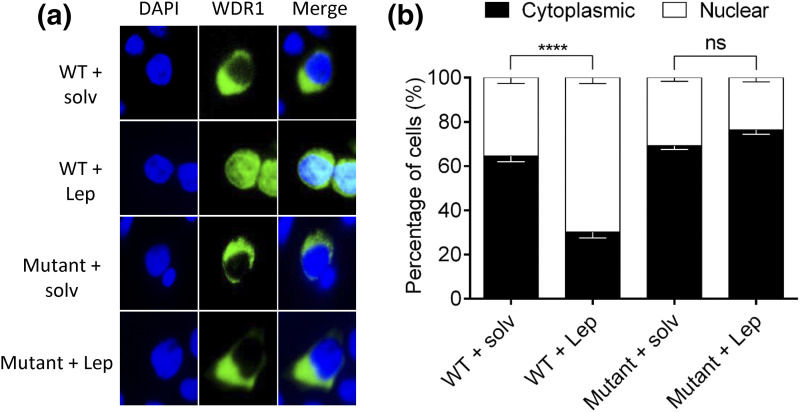
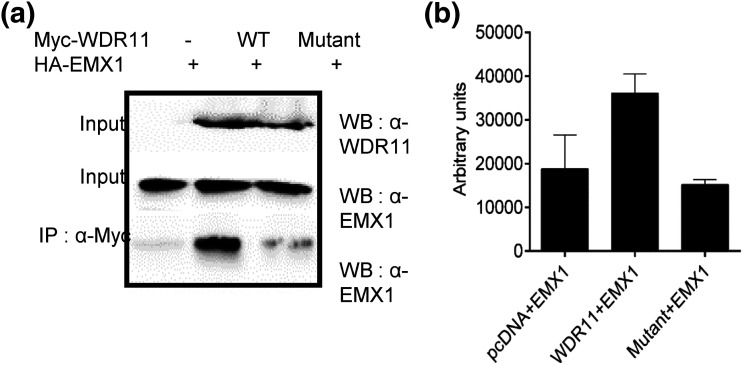
Whole exome sequencing (WES) was performed by using standard techniques, with prioritized genetic variants confirmed via Sanger sequencing. To investigate the effects of one candidate variant on mutant WDR11 function, Western blotting and coimmunofluorescence were used to assess binding capacity, and leptomycin B exposure along with immunofluorescence was used to assess nuclear localization.
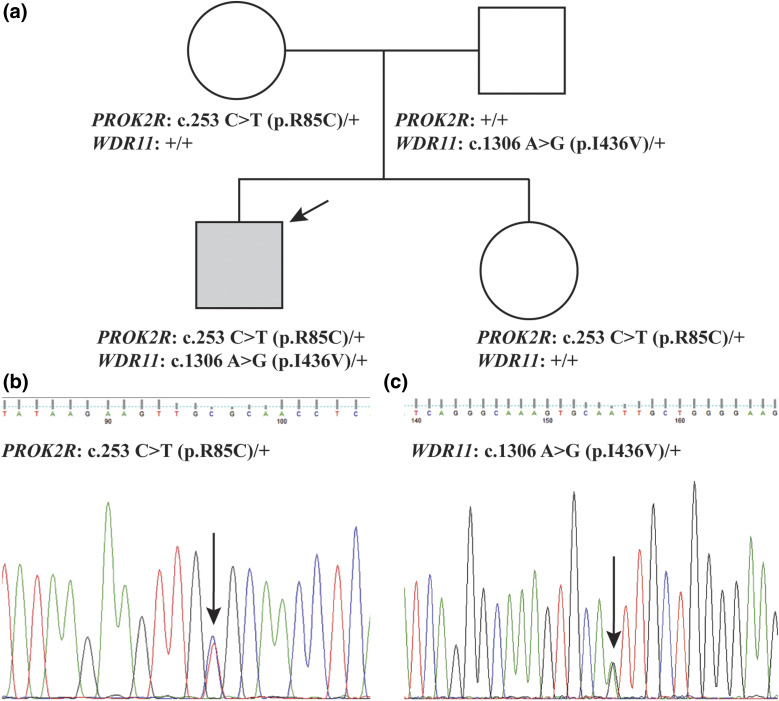
We describe a child who presented in infancy with combined pituitary hormone deficiencies and whose brain imaging demonstrated a small anterior pituitary, ectopic posterior pituitary, and a thin, interrupted stalk. WES demonstrated heterozygous missense mutations in two genes required for pituitary development, a known loss-of-function mutation in PROKR2 (c.253C>T;p.R85C) inherited from an unaffected mother, and a WDR11 (c.1306A>G;p.I436V) mutation inherited from an unaffected father. Mutant WDR11 loses its capacity to bind to its functional partner, EMX1, and to localize to the nucleus.
WES in a child with PSIS and his unaffected family implicates a digenic mechanism of inheritance. In cases of hypopituitarism in which there is incomplete segregation of a monogenic genotype with the phenotype, the possibility that a second genetic locus is involved should be considered.
垂体柄中断综合征(PSIS,孤儿病编号:ORPHA95496)是一种垂体先天性缺陷疾病,其特征为垂体柄极细/中断、垂体后叶异位(或缺失)以及垂体前叶发育不全或未发育。人们越来越认识到这种疾病复杂的遗传模式。
本研究旨在确定一名患病儿童垂体柄中断综合征的遗传病因。
采用标准技术进行全外显子组测序(WES),通过桑格测序确认优先选择的基因变异。为了研究一个候选变异对突变型WDR11功能的影响,使用蛋白质免疫印迹法和共免疫荧光法评估结合能力,使用放线菌素B处理结合免疫荧光法评估核定位。
我们描述了一名婴儿期出现联合垂体激素缺乏的儿童,其脑部影像学检查显示垂体前叶小、垂体后叶异位以及垂体柄细且中断。全外显子组测序显示,在垂体发育所需的两个基因中存在杂合错义突变,一个是从未患病母亲遗传而来的已知功能丧失突变PROKR2(c.253C>T;p.R85C),另一个是从未患病父亲遗传而来的WDR11(c.1306A>G;p.I436V)突变。突变型WDR11失去了与功能伙伴EMX1结合并定位于细胞核的能力。
对一名患有垂体柄中断综合征的儿童及其未患病家庭成员进行全外显子组测序表明存在双基因遗传机制。在单基因基因型与表型不完全分离的垂体功能减退病例中,应考虑第二个基因位点参与的可能性。