Department of Ophthalmology, Indiana University, Indianapolis, IN, USA.
Department of Ophthalmology, Stanford University, Palo Alto, CA, USA.
Sci Rep. 2017 May 4;7(1):1442. doi: 10.1038/s41598-017-01447-3.
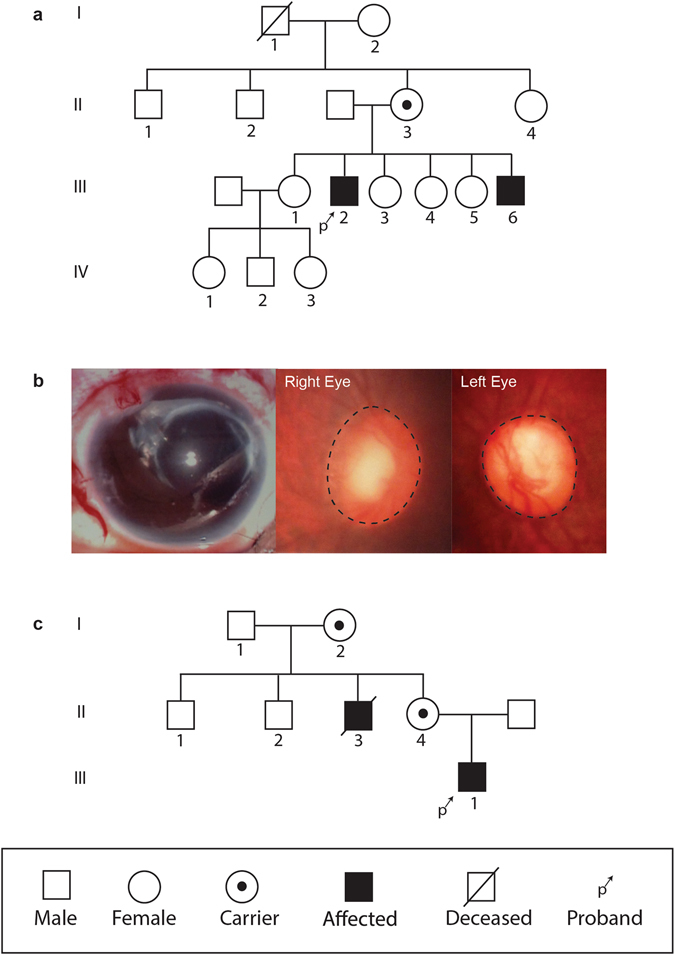
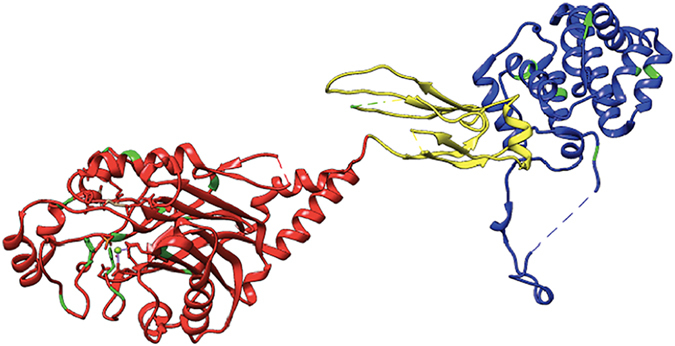
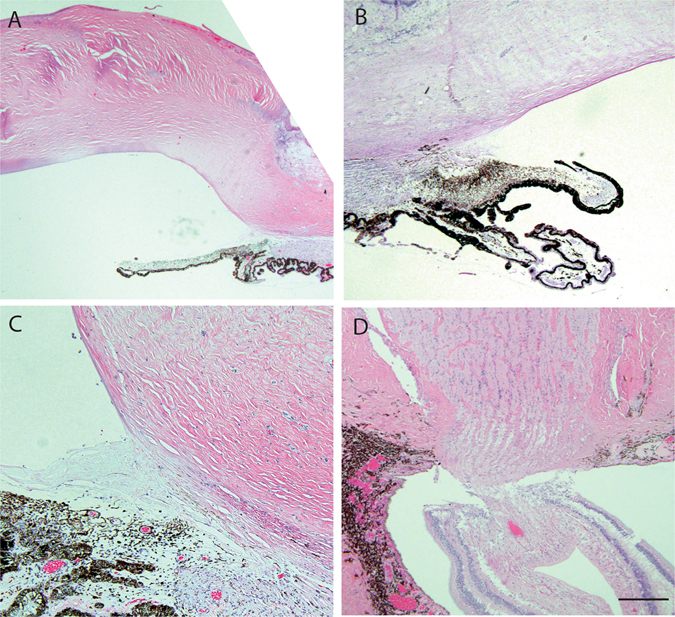
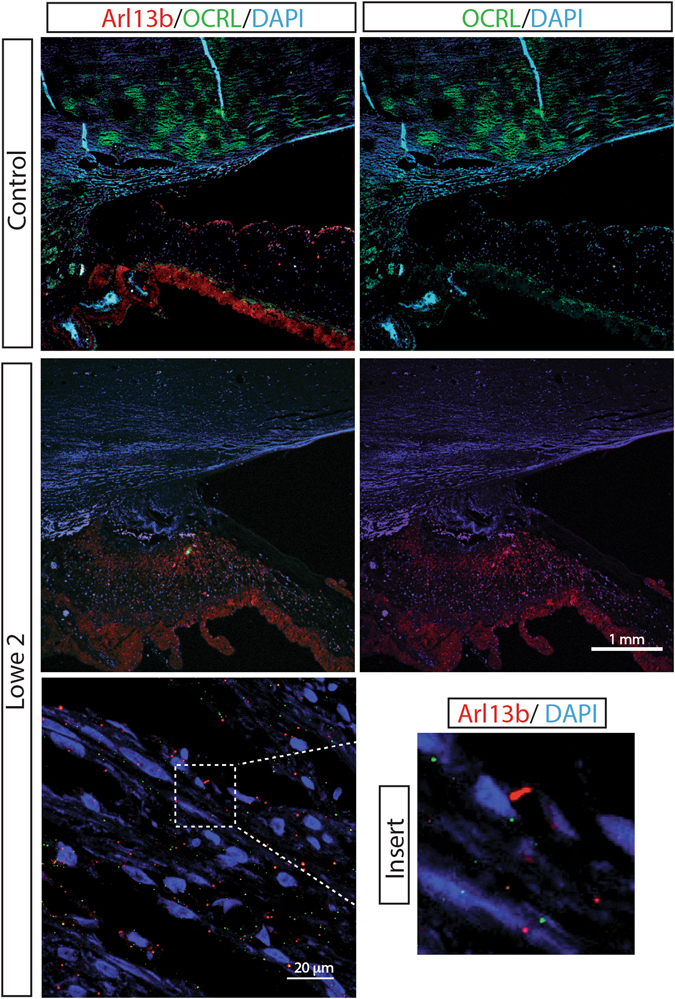
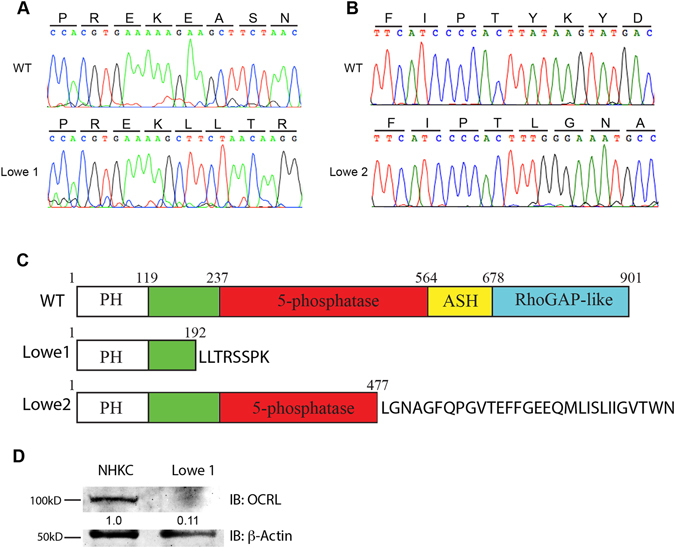
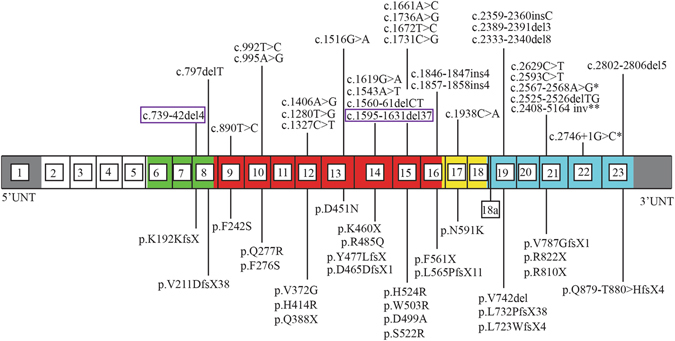
Mutations in the OCRL1 gene result in the oculocerebrorenal syndrome of Lowe, with symptoms including congenital bilateral cataracts, glaucoma, renal failure, and neurological impairments. OCRL1 encodes an inositol polyphosphate 5-phosphatase which preferentially dephosphorylates phosphatidylinositide 4,5 bisphosphate (PI(4,5)P). We have identified two novel mutations in two unrelated Lowe syndrome patients with congenital glaucoma. Novel deletion mutations are detected at c.739-742delAAAG in Lowe patient 1 and c.1595-1631del in Lowe patient 2. End stage glaucoma in patient 2 resulted in the enucleation of the eye, which on histology demonstrated corneal keloid, fibrous infiltration of the angle, ectropion uvea, retinal gliosis, and retinal ganglion cell loss. We measured OCRL protein levels in patient keratinocytes and found that Lowe 1 patient cells had significantly reduced OCRL protein as compared to the control keratinocytes. Genotype-phenotype correlation of OCRL1 mutations associated with congenital glaucoma revealed clustering of missense and deletion mutations in the 5-phosphatase domain and the RhoGAP-like domain. In conclusion, we report novel OCRL1 mutations in Lowe syndrome patients and the corresponding histopathologic analysis of one patient's ocular pathology.
OCRL1 基因突变导致 Lowe 眼-脑-肾综合征,其症状包括先天性双侧白内障、青光眼、肾衰竭和神经损伤。OCRL1 编码一种肌醇多磷酸 5-磷酸酶,它优先去磷酸化磷脂酰肌醇 4,5 二磷酸 (PI(4,5)P)。我们在两名先天性青光眼 Lowe 综合征患者中发现了两个新的突变。在 Lowe 患者 1 中检测到 c.739-742delAAAG 的新缺失突变,在 Lowe 患者 2 中检测到 c.1595-1631del 的缺失突变。患者 2 的晚期青光眼导致眼球摘除,组织学显示角膜瘢痕、角纤维浸润、葡萄膜外翻、视网膜胶质增生和视网膜神经节细胞丧失。我们测量了患者角质形成细胞中的 OCRL 蛋白水平,发现 Lowe 1 患者细胞的 OCRL 蛋白水平明显低于对照角质形成细胞。与先天性青光眼相关的 OCRL1 突变的基因型-表型相关性显示 5-磷酸酶结构域和 RhoGAP 样结构域的错义和缺失突变聚集。总之,我们报道了 Lowe 综合征患者中 OCRL1 的新突变,并对一名患者的眼部病变进行了相应的组织病理学分析。