Kawamata Hibiki, Peixoto Pablo, Konrad Csaba, Palomo Gloria, Bredvik Kirsten, Gerges Meri, Valsecchi Federica, Petrucelli Leonard, Ravits John M, Starkov Anatoly, Manfredi Giovanni
Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, 407 East 61st Street, RR507, New York, NY, 10065, USA.
Department of Natural Sciences, CUNY Baruch College, New York, NY, USA.
Mol Neurodegener. 2017 May 8;12(1):37. doi: 10.1186/s13024-017-0180-1.
Mitochondrial dysfunction has been linked to the pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Functional studies of mitochondrial bioenergetics have focused mostly on superoxide dismutase 1 (SOD1) mutants, and showed that mutant human SOD1 impairs mitochondrial oxidative phosphorylation, calcium homeostasis, and dynamics. However, recent reports have indicated that alterations in transactivation response element DNA-binding protein 43 (TDP-43) can also lead to defects of mitochondrial morphology and dynamics. Furthermore, it was proposed that TDP-43 mutations cause oxidative phosphorylation impairment associated with respiratory chain defects and that these effects were caused by mitochondrial localization of the mutant protein. Here, we investigated the presence of bioenergetic defects in the brain of transgenic mice expressing human mutant TDP-43 (TDP-43 mice), patient derived fibroblasts, and human cells expressing mutant forms of TDP-43.
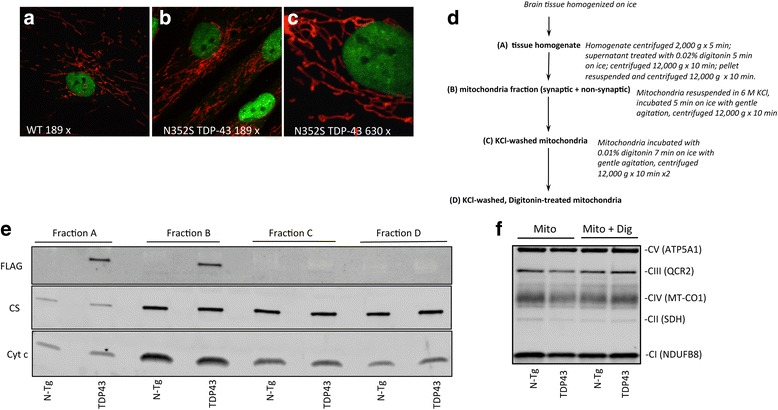
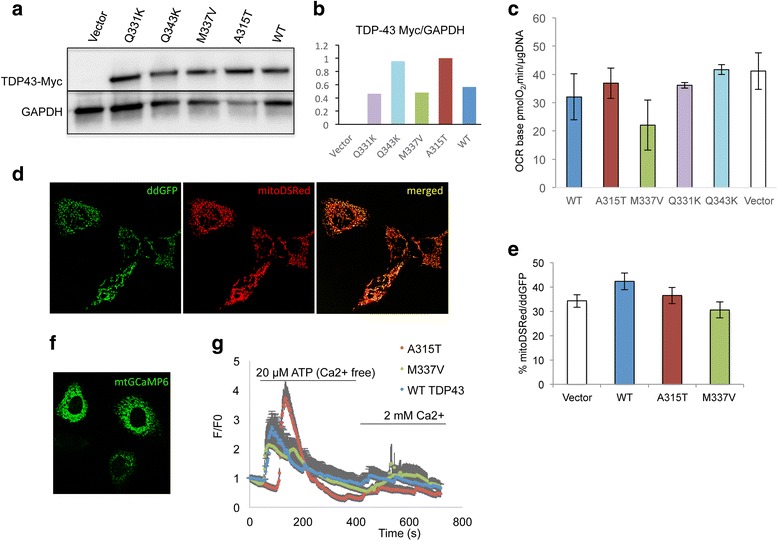
In the brain of TDP-43 mice, TDP-43 mutant fibroblasts, and cells expressing mutant TDP-43, we tested several bioenergetics parameters, including mitochondrial respiration, ATP synthesis, and calcium handling. Differences between mutant and control samples were evaluated by student t-test or by ANOVA, followed by Bonferroni correction, when more than two groups were compared. Mitochondrial localization of TDP-43 was investigated by immunocytochemistry in fibroblasts and by subcellular fractionation and western blot of mitochondrial fractions in mouse brain.
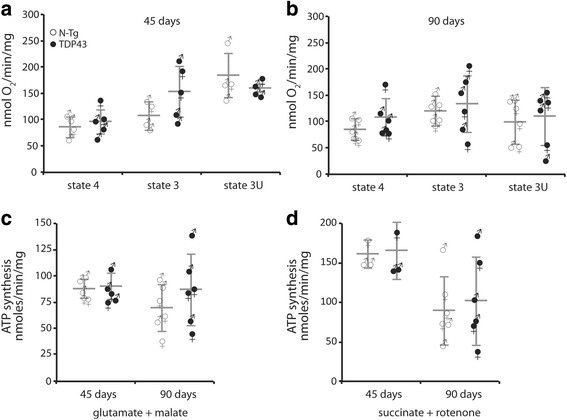
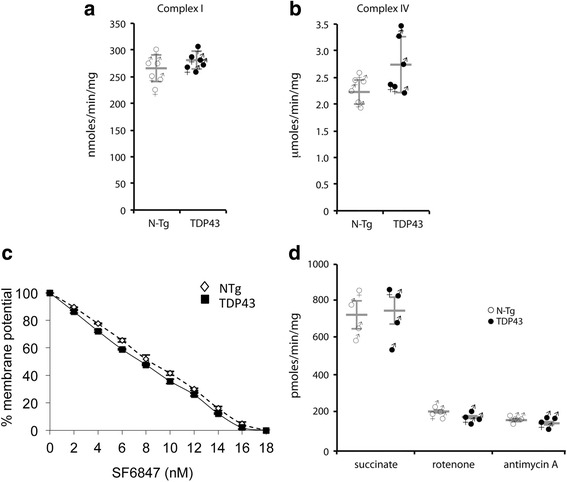
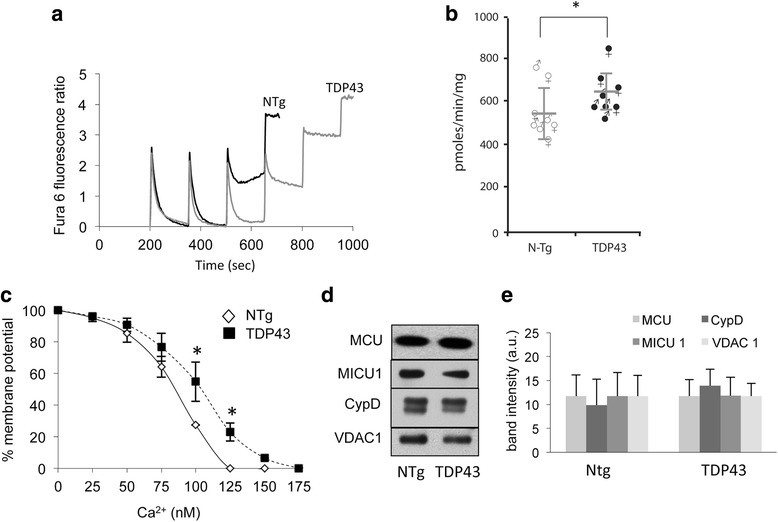
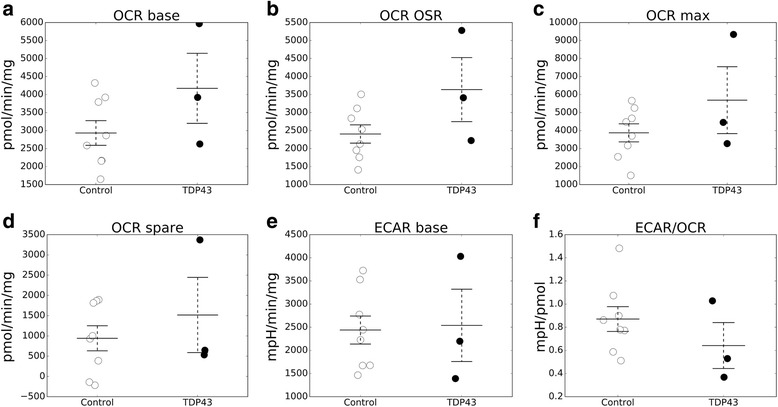
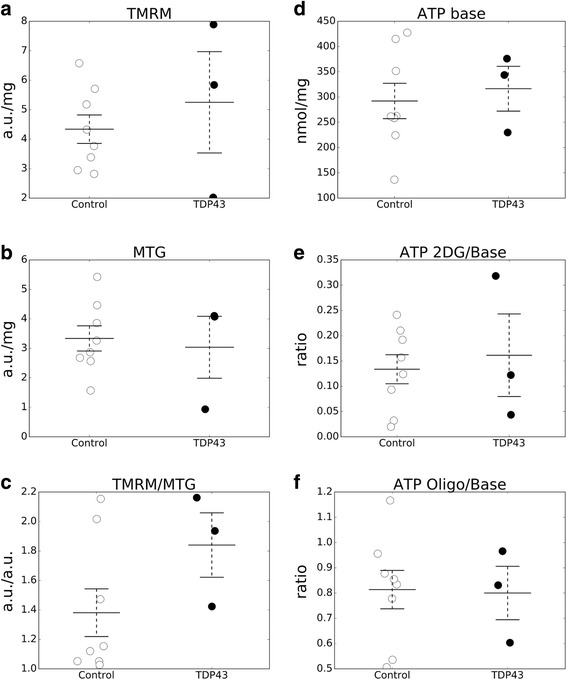
We did not observe defects in any of the mitochondrial bioenergetic functions that were tested in TDP-43 mutants. We detected a small amount of TDP-43 peripherally associated with brain mitochondria. However, there was no correlation between TDP-43 associated with mitochondria and respiratory chain dysfunction. In addition, we observed increased calcium uptake in mitochondria from TDP-43 mouse brain and cells expressing A315T mutant TDP-43.
While alterations of mitochondrial morphology and dynamics in TDP-43 mutant neurons are well established, the present study did not demonstrate oxidative phosphorylation defects in TDP-43 mutants, in vitro and in vivo. On the other hand, the increase in mitochondrial calcium uptake in A315T TDP-43 mutants was an intriguing finding, which needs to be investigated further to understand its mechanisms and potential pathogenic implications.
线粒体功能障碍与肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTLD)的发病机制有关。线粒体生物能量学的功能研究主要集中在超氧化物歧化酶1(SOD1)突变体上,并表明突变的人类SOD1会损害线粒体氧化磷酸化、钙稳态和动力学。然而,最近的报道表明,反式激活应答元件DNA结合蛋白43(TDP-43)的改变也会导致线粒体形态和动力学缺陷。此外,有人提出TDP-43突变会导致与呼吸链缺陷相关的氧化磷酸化损伤,并且这些效应是由突变蛋白的线粒体定位引起的。在此,我们研究了表达人类突变TDP-43的转基因小鼠(TDP-43小鼠)大脑、患者来源的成纤维细胞以及表达TDP-43突变形式的人类细胞中生物能量缺陷的存在情况。
在TDP-43小鼠的大脑、TDP-43突变成纤维细胞以及表达突变TDP-43的细胞中,我们测试了几个生物能量学参数,包括线粒体呼吸、ATP合成和钙处理。当比较两组以上时,通过学生t检验或方差分析评估突变样本和对照样本之间的差异,然后进行Bonferroni校正。通过免疫细胞化学在成纤维细胞中以及通过亚细胞分级分离和小鼠脑线粒体分级分离的蛋白质印迹法研究TDP-43的线粒体定位。
我们在TDP-43突变体中测试的任何线粒体生物能量功能中均未观察到缺陷。我们检测到少量TDP-43在外周与脑线粒体相关。然而,与线粒体相关的TDP-43与呼吸链功能障碍之间没有相关性。此外,我们观察到来自TDP-43小鼠脑和表达A315T突变TDP-43的细胞的线粒体中钙摄取增加。
虽然TDP-43突变神经元中线粒体形态和动力学的改变已得到充分证实,但本研究在体外和体内均未证明TDP-43突变体存在氧化磷酸化缺陷。另一方面,A315T TDP-43突变体中线粒体钙摄取的增加是一个有趣的发现,需要进一步研究以了解其机制和潜在的致病意义。