Lester & Sue Smith Breast Center, Baylor College of Medicine, Houston, Texas.
Dan L. Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, Texas.
Clin Cancer Res. 2017 Sep 1;23(17):5123-5134. doi: 10.1158/1078-0432.CCR-16-2191. Epub 2017 May 9.
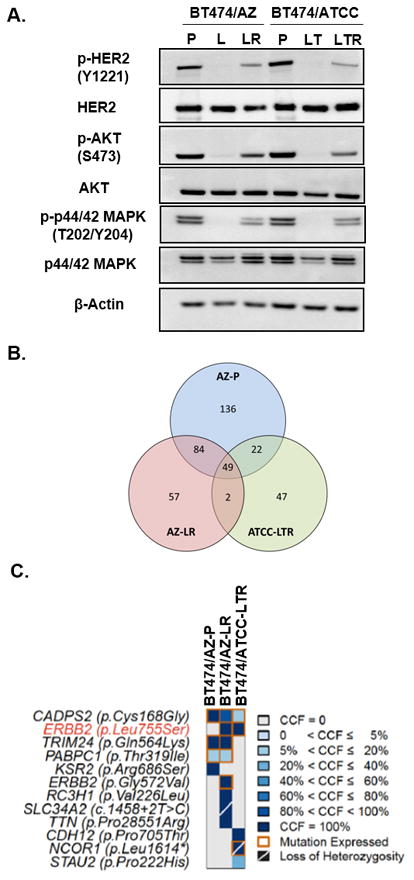
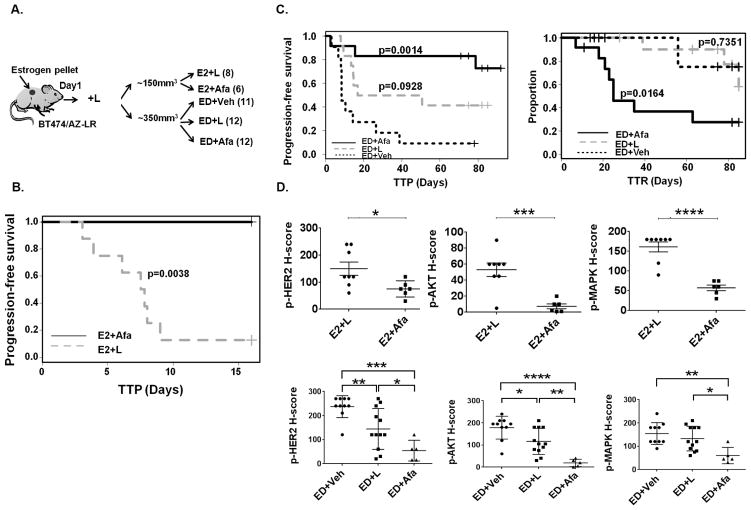
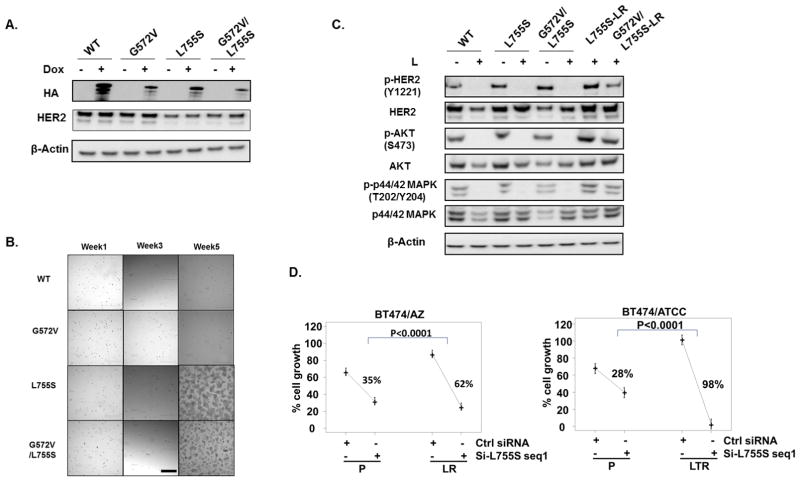
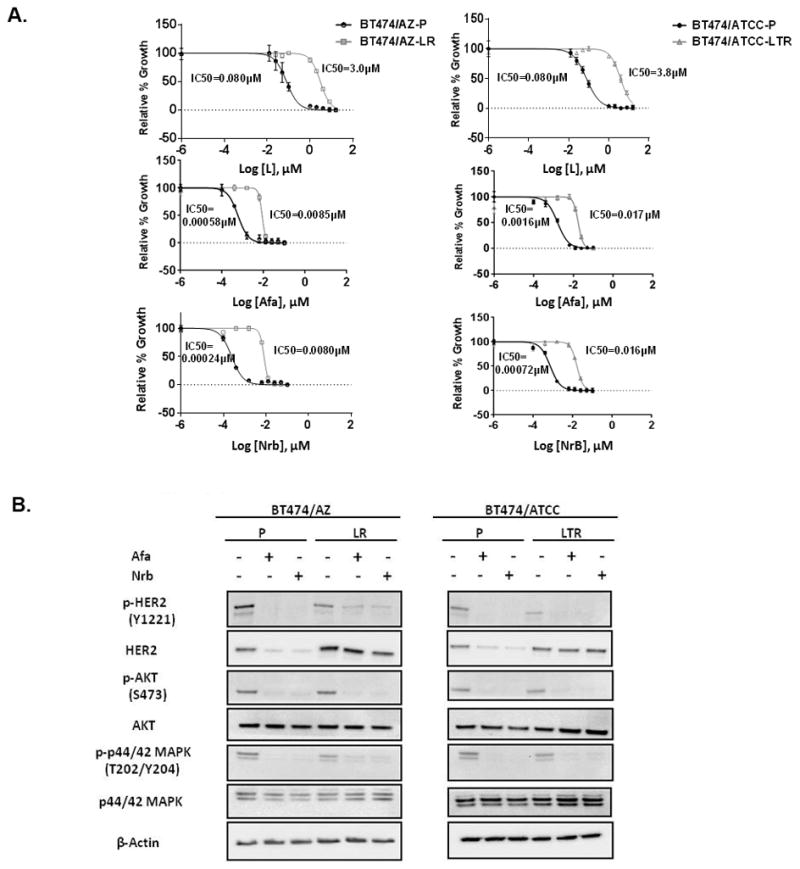
Resistance to anti-HER2 therapies in HER2 breast cancer can occur through activation of alternative survival pathways or reactivation of the HER signaling network. Here we employed BT474 parental and treatment-resistant cell line models to investigate a mechanism by which HER2 breast cancer can reactivate the HER network under potent HER2-targeted therapies. Resistant derivatives to lapatinib (L), trastuzumab (T), or the combination (LR/TR/LTR) were developed independently from two independent estrogen receptor ER/HER2 BT474 cell lines (AZ/ATCC). Two derivatives resistant to the lapatinib-containing regimens (BT474/AZ-LR and BT474/ATCC-LTR lines) that showed HER2 reactivation at the time of resistance were subjected to massive parallel sequencing and compared with parental lines. Ectopic expression and mutant-specific siRNA interference were applied to analyze the mutation functionally. and experiments were performed to test alternative therapies for mutant HER2 inhibition. Genomic analyses revealed that the L755S mutation was the only common somatic mutation gained in the BT474/AZ-LR and BT474/ATCC-LTR lines. Ectopic expression of L755S induced acquired lapatinib resistance in the BT474/AZ, SK-BR-3, and AU565 parental cell lines. L755S-specific siRNA knockdown reversed the resistance in BT474/AZ-LR and BT474/ATCC-LTR lines. The HER1/2-irreversible inhibitors afatinib and neratinib substantially inhibited both resistant cell growth and the HER2 and downstream AKT/MAPK signaling driven by L755S and HER2 reactivation through acquisition of the L755S mutation was identified as a mechanism of acquired resistance to lapatinib-containing HER2-targeted therapy in preclinical HER2-amplified breast cancer models, which can be overcome by irreversible HER1/2 inhibitors. .
曲妥珠单抗耐药的 HER2 阳性乳腺癌的产生可能是由于激活了替代的生存途径,或者是重新激活了 HER 信号网络。在此,我们采用 BT474 亲本细胞和耐药细胞系模型来研究一种机制,即 HER2 阳性乳腺癌在受到强力的 HER2 靶向治疗后,如何重新激活 HER 网络。从两个独立的雌激素受体 ER/HER2 BT474 细胞系(AZ/ATCC)中,我们独立开发了对拉帕替尼(L)、曲妥珠单抗(T)或联合用药(LR/TR/LTR)耐药的耐药衍生物。两个对含有拉帕替尼的方案耐药的衍生物(BT474/AZ-LR 和 BT474/ATCC-LTR 株)在耐药时表现出 HER2 重新激活,我们对其进行了大规模平行测序,并与亲本株进行了比较。通过异位表达和突变体特异性 siRNA 干扰来分析突变的功能。进行了 和 实验来测试用于抑制突变型 HER2 的替代治疗方法。基因组分析显示,L755S 突变是 BT474/AZ-LR 和 BT474/ATCC-LTR 株中唯一获得的共同体细胞突变。L755S 的异位表达在 BT474/AZ、SK-BR-3 和 AU565 亲本细胞系中诱导了获得性拉帕替尼耐药。L755S 特异性 siRNA 敲低逆转了 BT474/AZ-LR 和 BT474/ATCC-LTR 株的耐药性。HER1/2 不可逆抑制剂阿法替尼和奈拉替尼通过获得 L755S 突变,显著抑制了耐药细胞的生长以及由 L755S 和 HER2 重新激活驱动的下游 AKT/MAPK 信号,在临床前 HER2 扩增乳腺癌模型中鉴定出获得性拉帕替尼耐药的机制是对包含曲妥珠单抗的 HER2 靶向治疗的获得性耐药,该耐药性可以被不可逆的 HER1/2 抑制剂克服。