Chen Yating, Luo Peng, Li Zhongli, Hu Hengping, Wu Duobin, Xu Tingting, Wang Xingzuo, Xie Haiting
Department of Neurology, Zhujiang Hospital of Southern Medical University, Guangzhou, China.
Medicine (Baltimore). 2017 May;96(19):e6792. doi: 10.1097/MD.0000000000006792.
Kennedy disease (KD) is also known as spinal bulbar muscular dystrophy. As KD has similar symptoms with most neuromuscular diseases, so it is difficult to make a rapid diagnosis clinically.
We report a case of a 43-year-old male with progressive limb proximal weakness without family history. Physical examination showed gynecomastia, erectile dysfunction, bilateral tendon reflex and quadriceps weakness, and tongue muscle atrophy.
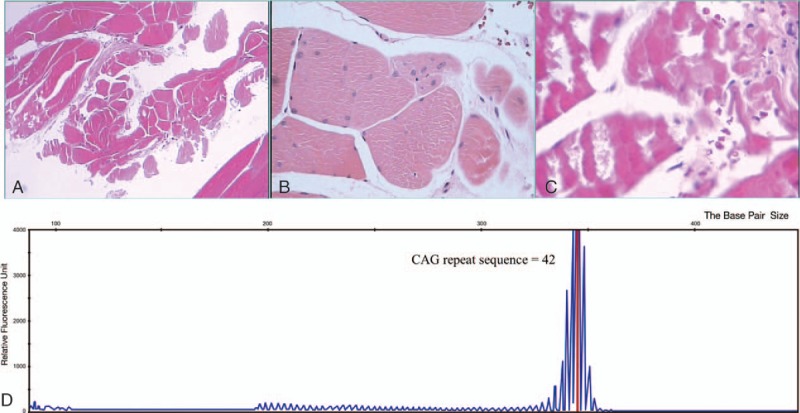
Laboratory examination found increased creatine kinase, impaired glucose tolerance, and abnormal lactic acid values. There was no mutation or copy number variant in SMN1 gene and related mitochondrion genes tested, even with the use of multiplex ligation probe- dependent amplification technique. Diagnosis was confirmed with genetic analysis which displayed trinucleotide CAG (glutamine)- repeat expansion in the androgen-receptor gene.
The patient achieved good prognosis with symptomatic treatment after diagnosis.
To diagnose KD, clinicians should pay more attention to differentiate KD and myasthenia gravis, mitochondrial myopathy, and amyotrophic lateral sclerosis. Gene analysis was the key in detecting this rare confusing disease in the patient.
肯尼迪病(KD)也被称为脊髓延髓肌肉萎缩症。由于KD与大多数神经肌肉疾病有相似症状,因此临床上难以快速做出诊断。
我们报告一例43岁男性患者,其渐进性肢体近端无力,无家族病史。体格检查显示有男性乳房发育、勃起功能障碍、双侧腱反射和股四头肌无力以及舌肌萎缩。
实验室检查发现肌酸激酶升高、葡萄糖耐量受损和乳酸值异常。即使使用多重连接探针依赖扩增技术,所检测的SMN1基因和相关线粒体基因也未发现突变或拷贝数变异。通过基因分析确诊,该分析显示雄激素受体基因中有三核苷酸CAG(谷氨酰胺)重复扩增。
患者诊断后经对症治疗预后良好。
为诊断KD,临床医生应更加注意鉴别KD与重症肌无力、线粒体肌病和肌萎缩侧索硬化症。基因分析是检测该患者这种罕见疑难疾病的关键。