Zhou Shiyong, Liu Pengfei, Zhang Huilai
Department of Lymphoma, Sino‑US Center of Lymphoma and Leukemia, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin's Clinical Research Center for Cancer, Tianjin 300060, P.R. China.
Mol Med Rep. 2017 Jul;16(1):281-287. doi: 10.3892/mmr.2017.6581. Epub 2017 May 12.
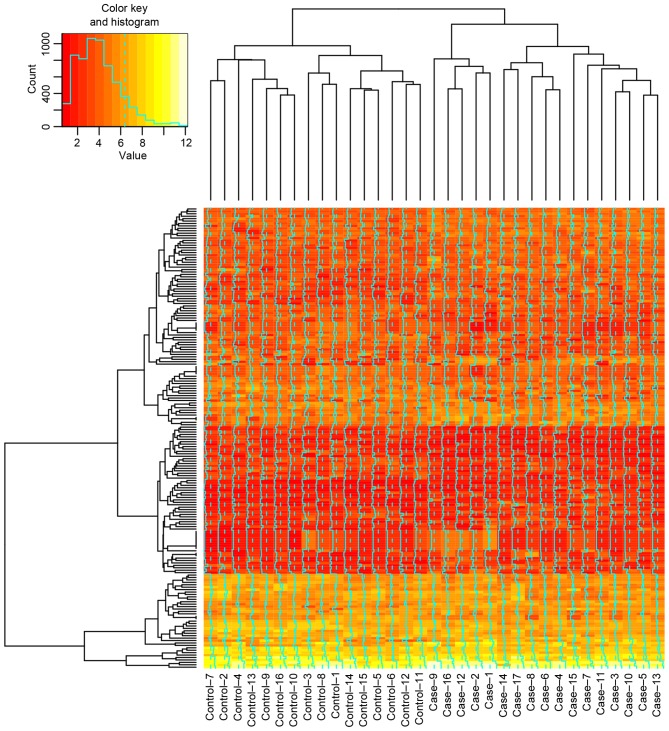
Acute myeloid leukemia (AML) is a frequently occurring malignant disease of the blood and may result from a variety of genetic disorders. The present study aimed to identify the underlying mechanisms associated with the therapeutic effects of decitabine and cytarabine on AML, using microarray analysis. The microarray datasets GSE40442 and GSE40870 were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) and differentially methylated sites were identified in AML cells treated with decitabine compared with those treated with cytarabine via the Linear Models for Microarray Data package, following data pre‑processing. Gene Ontology (GO) analysis of DEGs was performed using the Database for Annotation, Visualization and Integrated Analysis Discovery. Genes corresponding to the differentially methylated sites were obtained using the annotation package of the methylation microarray platform. The overlapping genes were identified, which exhibited the opposite variation trend between gene expression and DNA methylation. Important transcription factor (TF)‑gene pairs were screened out, and a regulated network subsequently constructed. A total of 190 DEGs and 540 differentially methylated sites were identified in AML cells treated with decitabine compared with those treated with cytarabine. A total of 36 GO terms of DEGs were enriched, including nucleosomes, protein‑DNA complexes and the nucleosome assembly. The 540 differentially methylated sites were located on 240 genes, including the acid‑repeat containing protein (ACRC) gene that was additionally differentially expressed. In addition, 60 TF pairs and overlapped methylated sites, and 140 TF‑pairs and DEGs were screened out. The regulated network included 68 nodes and 140 TF‑gene pairs. The present study identified various genes including ACRC and proliferating cell nuclear antigen, in addition to various TFs, including TATA‑box binding protein associated factor 1 and CCCTC‑binding factor, which may be potential therapeutic targets of AML.
急性髓系白血病(AML)是一种常见的血液恶性疾病,可能由多种基因紊乱引起。本研究旨在通过微阵列分析确定地西他滨和阿糖胞苷对AML治疗效果的潜在机制。从基因表达综合数据库下载了微阵列数据集GSE40442和GSE40870。在数据预处理后,通过微阵列数据线性模型软件包,鉴定了地西他滨处理的AML细胞与阿糖胞苷处理的AML细胞相比的差异表达基因(DEG)和差异甲基化位点。使用注释、可视化和综合分析发现数据库对DEG进行基因本体(GO)分析。使用甲基化微阵列平台的注释软件包获得与差异甲基化位点对应的基因。鉴定出重叠基因,其在基因表达和DNA甲基化之间呈现相反的变化趋势。筛选出重要的转录因子(TF)-基因对,随后构建调控网络。与阿糖胞苷处理的AML细胞相比,地西他滨处理的AML细胞中共鉴定出190个DEG和540个差异甲基化位点。共富集了36个DEG的GO术语,包括核小体、蛋白质-DNA复合物和核小体组装。540个差异甲基化位点位于240个基因上,包括另外差异表达的含酸重复蛋白(ACRC)基因。此外,筛选出60个TF对和重叠甲基化位点,以及140个TF-对和DEG。调控网络包括68个节点和140个TF-基因对。本研究鉴定出包括ACRC和增殖细胞核抗原在内的各种基因,以及包括TATA框结合蛋白相关因子1和CCCTC结合因子在内的各种TF,它们可能是AML的潜在治疗靶点。