Mahmood Khalid, Jung Chol-Hee, Philip Gayle, Georgeson Peter, Chung Jessica, Pope Bernard J, Park Daniel J
Melbourne Bioinformatics, The University of Melbourne, Melbourne, Australia.
Hum Genomics. 2017 May 16;11(1):10. doi: 10.1186/s40246-017-0104-8.
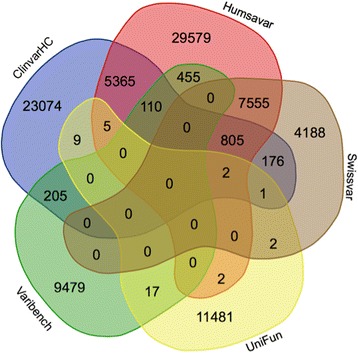
Genetic variant effect prediction algorithms are used extensively in clinical genomics and research to determine the likely consequences of amino acid substitutions on protein function. It is vital that we better understand their accuracies and limitations because published performance metrics are confounded by serious problems of circularity and error propagation. Here, we derive three independent, functionally determined human mutation datasets, UniFun, BRCA1-DMS and TP53-TA, and employ them, alongside previously described datasets, to assess the pre-eminent variant effect prediction tools.
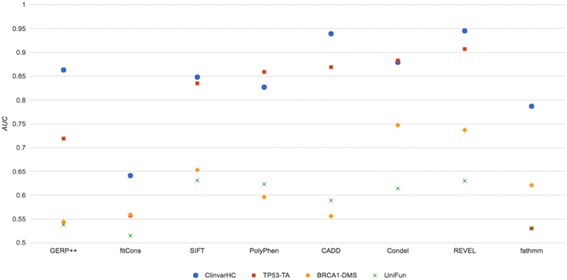
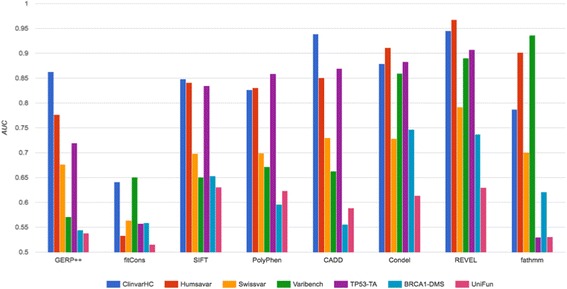
Apparent accuracies of variant effect prediction tools were influenced significantly by the benchmarking dataset. Benchmarking with the assay-determined datasets UniFun and BRCA1-DMS yielded areas under the receiver operating characteristic curves in the modest ranges of 0.52 to 0.63 and 0.54 to 0.75, respectively, considerably lower than observed for other, potentially more conflicted datasets.
These results raise concerns about how such algorithms should be employed, particularly in a clinical setting. Contemporary variant effect prediction tools are unlikely to be as accurate at the general prediction of functional impacts on proteins as reported prior. Use of functional assay-based datasets that avoid prior dependencies promises to be valuable for the ongoing development and accurate benchmarking of such tools.
基因变异效应预测算法在临床基因组学和研究中被广泛用于确定氨基酸替换对蛋白质功能可能产生的后果。我们必须更好地了解它们的准确性和局限性,因为已发表的性能指标受到严重的循环性和误差传播问题的混淆。在这里,我们推导了三个独立的、功能确定的人类突变数据集,即UniFun、BRCA1-DMS和TP53-TA,并将它们与先前描述的数据集一起用于评估卓越的变异效应预测工具。
变异效应预测工具的表观准确性受到基准数据集的显著影响。使用通过实验确定的数据集UniFun和BRCA1-DMS进行基准测试时,受试者工作特征曲线下的面积分别在0.52至0.63和0.54至0.75的适度范围内,远低于在其他可能更具冲突性的数据集上观察到的结果。
这些结果引发了对如何使用此类算法的担忧,尤其是在临床环境中。当代变异效应预测工具在对蛋白质功能影响的一般预测方面不太可能像之前报道的那样准确。使用避免先前依赖性的基于功能测定的数据集有望对这类工具的持续开发和准确基准测试有价值。