Ran Di, Daye Z John
Mel and Enid Zuckerman College of Public Health, The University of Arizona, Tucson, AZ 85724, USA.
Independent Researcher, Raleigh, NC 27612, USA.
Nucleic Acids Res. 2017 Jul 27;45(13):e127. doi: 10.1093/nar/gkx456.
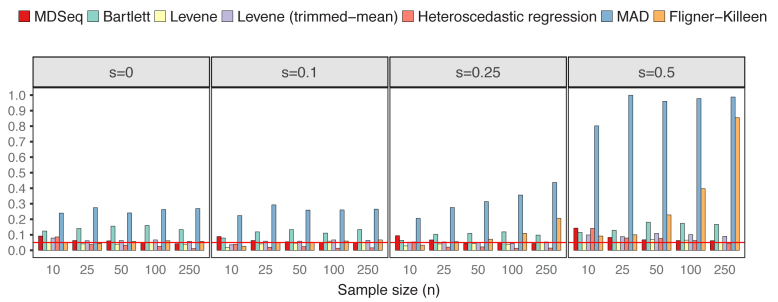
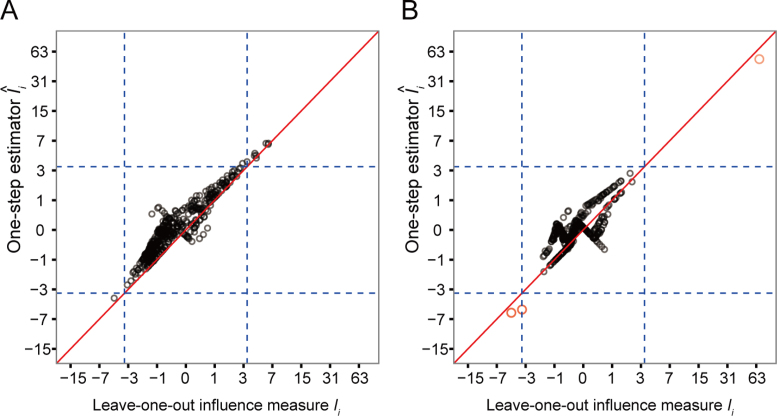
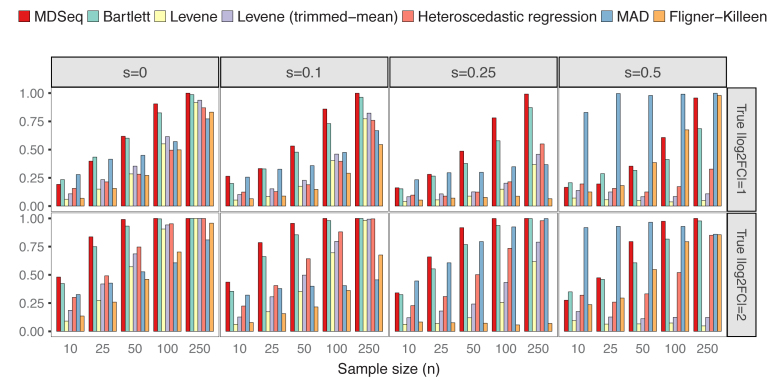
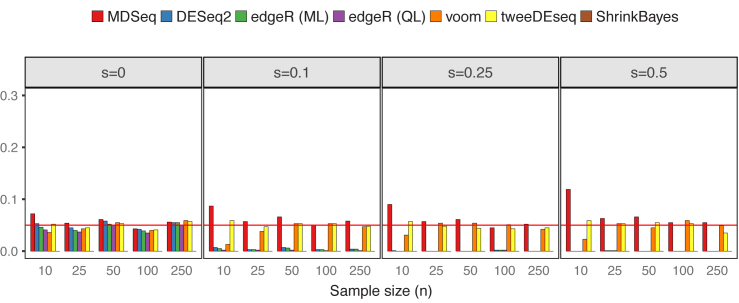
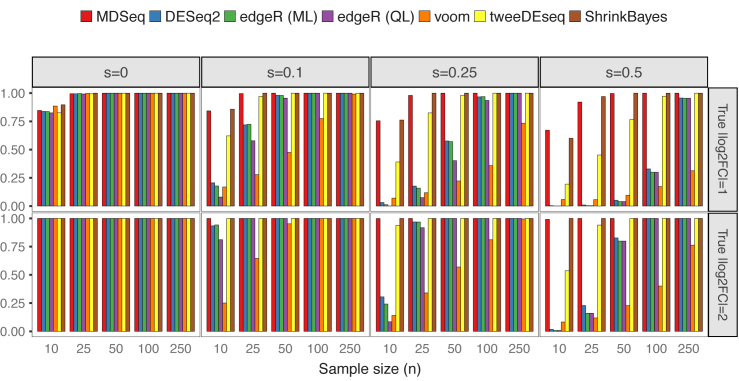
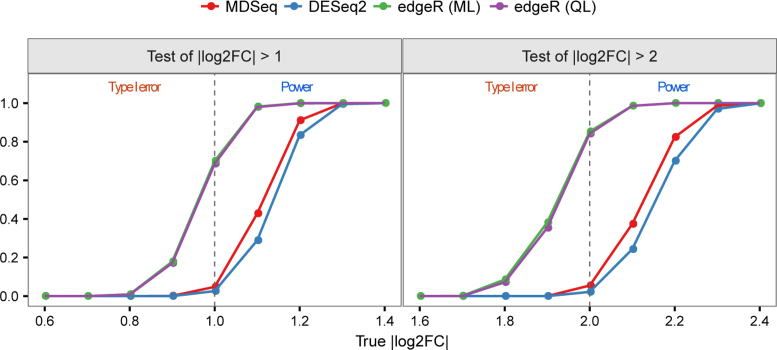
Rapidly decreasing cost of next-generation sequencing has led to the recent availability of large-scale RNA-seq data, that empowers the analysis of gene expression variability, in addition to gene expression means. In this paper, we present the MDSeq, based on the coefficient of dispersion, to provide robust and computationally efficient analysis of both gene expression means and variability on RNA-seq counts. The MDSeq utilizes a novel reparametrization of the negative binomial to provide flexible generalized linear models (GLMs) on both the mean and dispersion. We address challenges of analyzing large-scale RNA-seq data via several new developments to provide a comprehensive toolset that models technical excess zeros, identifies outliers efficiently, and evaluates differential expressions at biologically interesting levels. We evaluated performances of the MDSeq using simulated data when the ground truths are known. Results suggest that the MDSeq often outperforms current methods for the analysis of gene expression mean and variability. Moreover, the MDSeq is applied in two real RNA-seq studies, in which we identified functionally relevant genes and gene pathways. Specifically, the analysis of gene expression variability with the MDSeq on the GTEx human brain tissue data has identified pathways associated with common neurodegenerative disorders when gene expression means were conserved.
下一代测序成本的迅速下降使得大规模RNA测序数据近期得以获取,这不仅能分析基因表达均值,还能对基因表达变异性进行分析。在本文中,我们提出了基于离散系数的MDSeq,以对RNA测序计数中的基因表达均值和变异性进行稳健且计算高效的分析。MDSeq利用负二项式的一种新型重新参数化方法,在均值和离散度上提供灵活的广义线性模型(GLM)。我们通过几个新进展应对分析大规模RNA测序数据的挑战,以提供一个全面的工具集,该工具集能对技术上的过多零值进行建模、有效识别异常值,并在生物学上有意义的水平评估差异表达。当已知真实情况时,我们使用模拟数据评估了MDSeq的性能。结果表明,在分析基因表达均值和变异性方面,MDSeq常常优于当前方法。此外,MDSeq应用于两项真实的RNA测序研究,我们在其中鉴定出了功能相关的基因和基因通路。具体而言,在GTEx人类脑组织数据上使用MDSeq分析基因表达变异性时,当基因表达均值保持不变时,已鉴定出与常见神经退行性疾病相关的通路。