Institute of Mathematical Sciences, Universiti Malaya, Kuala Lumpur, Malaysia.
Universiti Malaya Centre for Data Analytics, Universiti Malaya, Kuala Lumpur, Malaysia.
PeerJ. 2023 Sep 29;11:e16126. doi: 10.7717/peerj.16126. eCollection 2023.
Pathological conditions may result in certain genes having expression variance that differs markedly from that of the control. Finding such genes from gene expression data can provide invaluable candidates for therapeutic intervention. Under the dominant paradigm for modeling RNA-Seq gene counts using the negative binomial model, tests of differential variability are challenging to develop, owing to dependence of the variance on the mean.
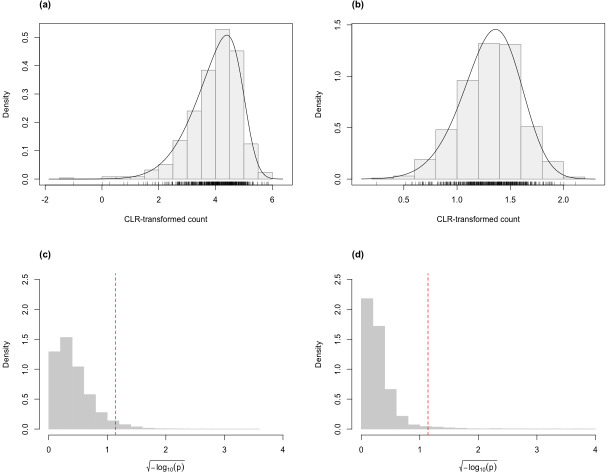
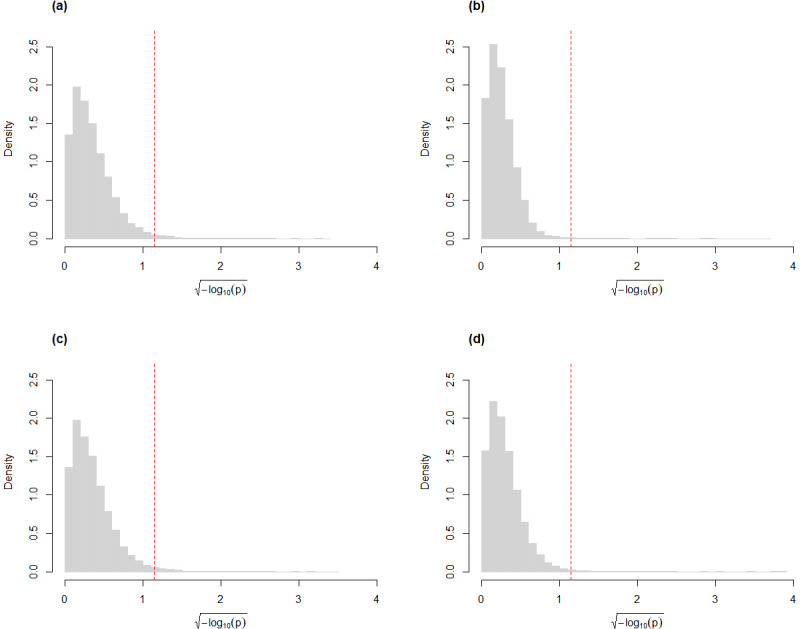
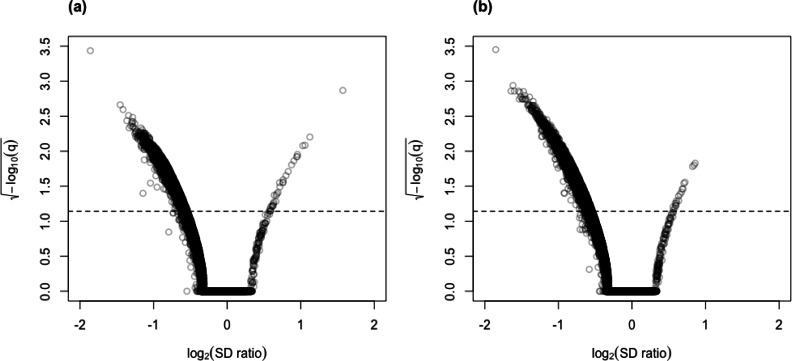
Here, we describe clrDV, a statistical method for detecting genes that show differential variability between two populations. We present the skew-normal distribution for modeling gene-wise null distribution of centered log-ratio transformation of compositional RNA-seq data.
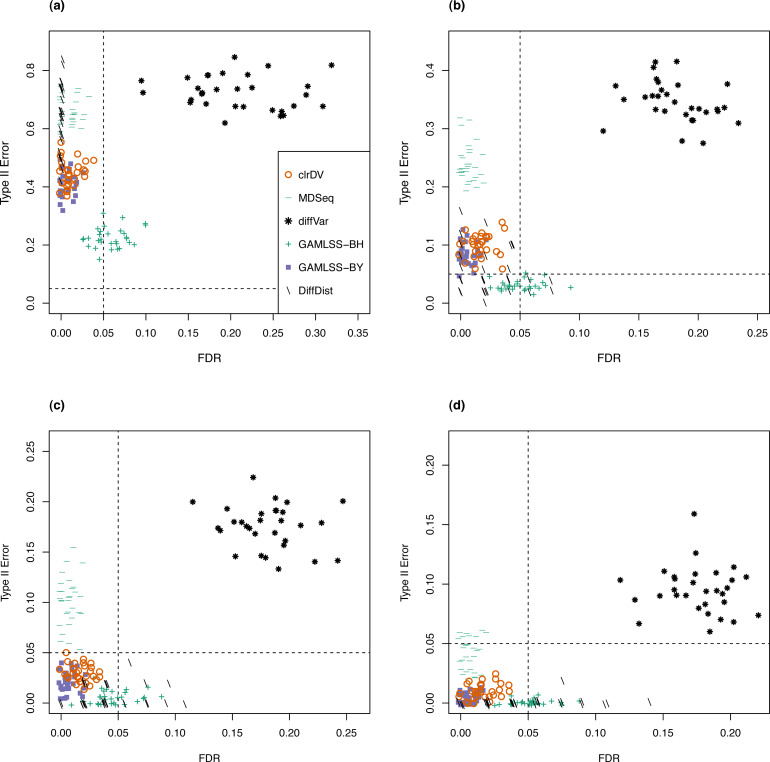
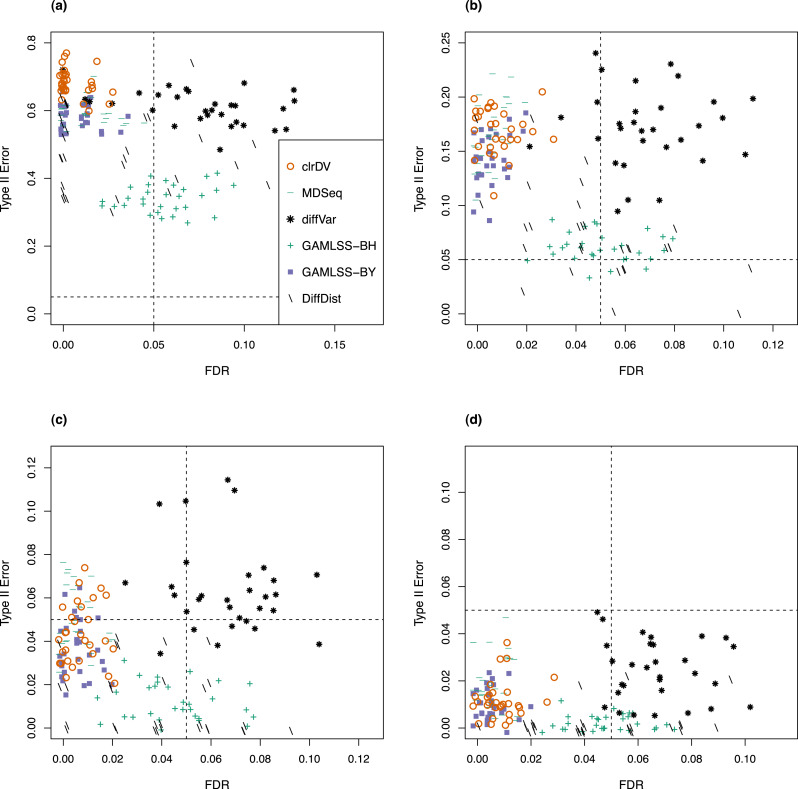
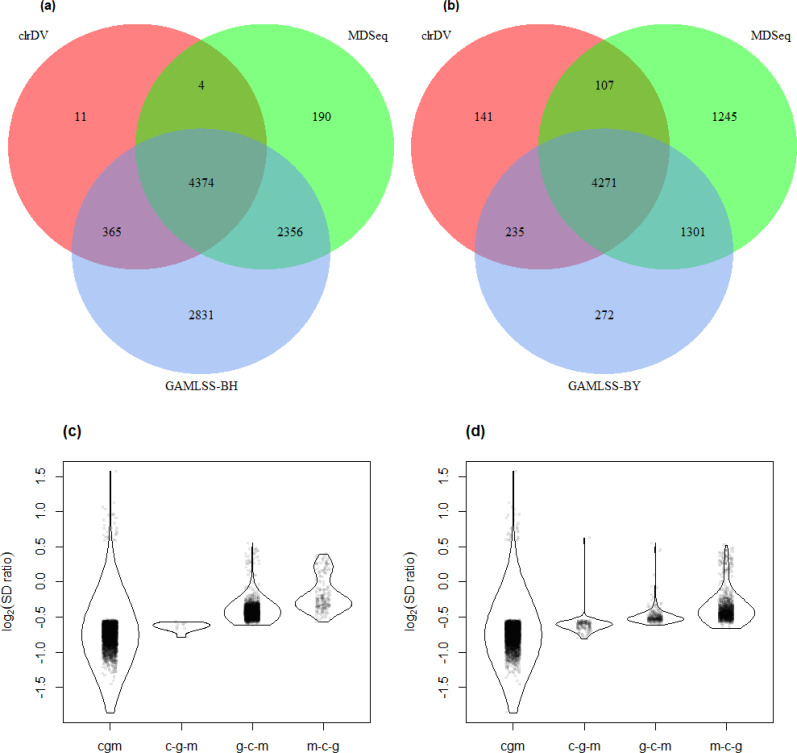
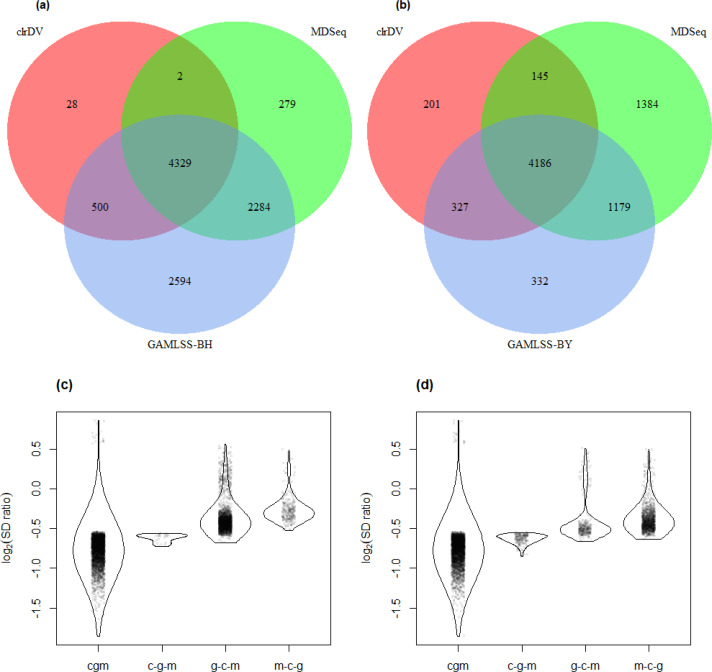
Simulation results show that clrDV has false discovery rate and probability of Type II error that are on par with or superior to existing methodologies. In addition, its run time is faster than its closest competitors, and remains relatively constant for increasing sample size per group. Analysis of a large neurodegenerative disease RNA-Seq dataset using clrDV successfully recovers multiple gene candidates that have been reported to be associated with Alzheimer's disease.
病理条件可能导致某些基因的表达方差与对照明显不同。从基因表达数据中找到这些基因,可以为治疗干预提供非常有价值的候选基因。在使用负二项模型对 RNA-Seq 基因计数进行建模的主流范例下,由于方差依赖于均值,因此很难开发出用于检测差异变异性的检验方法。
在这里,我们描述了 clrDV,这是一种用于检测两个群体之间差异变异性的基因的统计方法。我们提出了偏态正态分布,用于对基于中心对数比变换的组成性 RNA-seq 数据的基因进行零分布建模。
模拟结果表明,clrDV 的假发现率和第二类错误的概率与现有方法相当或优于现有方法。此外,它的运行时间比最接近的竞争对手快,并且随着每组样本量的增加而保持相对稳定。使用 clrDV 对大型神经退行性疾病 RNA-Seq 数据集进行分析,成功地恢复了多个已报道与阿尔茨海默病相关的基因候选物。