Park Jung Woo, Yan Li, Stoddard Chris, Wang Xiaofang, Yue Zhichao, Crandall Leann, Robinson Tiwanna, Chang Yuxiao, Denton Kyle, Li Enqin, Jiang Bin, Zhang Zhenwu, Martins-Taylor Kristen, Yee Siu-Pok, Nie Hong, Gu Feng, Si Wei, Xie Ting, Yue Lixia, Xu Ren-He
Faculty of Health Sciences, University of Macau, Taipa, Macau, China.
Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, Connecticut, USA.
Int J Biol Sci. 2017 Apr 10;13(5):588-603. doi: 10.7150/ijbs.19517. eCollection 2017.
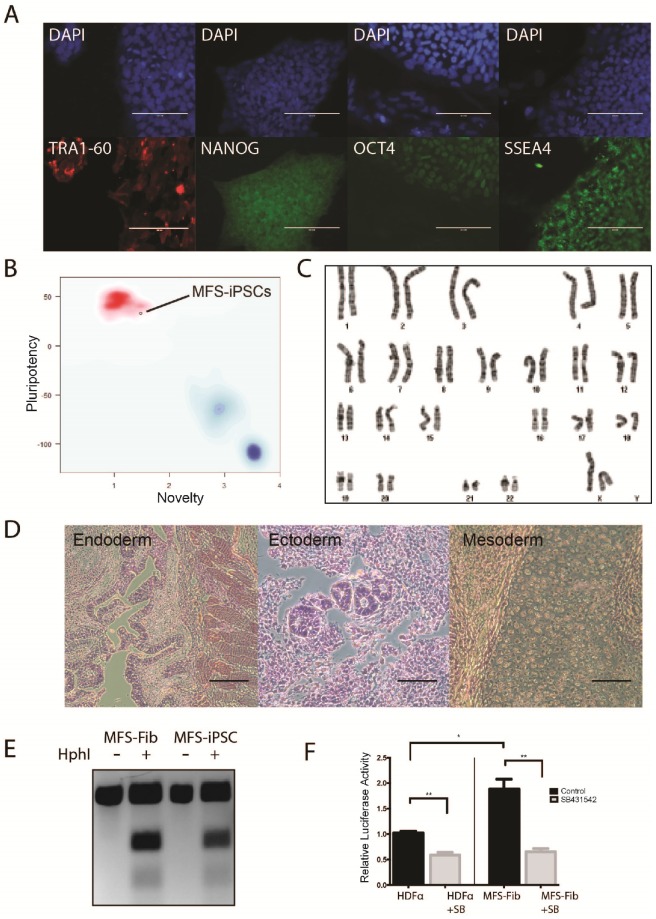
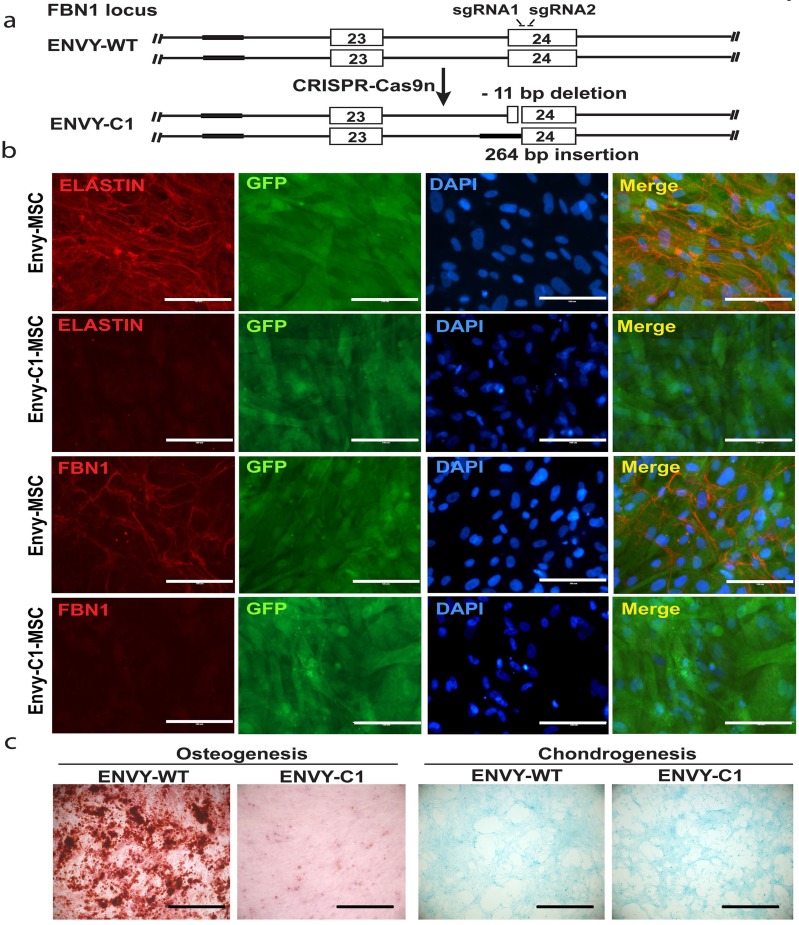
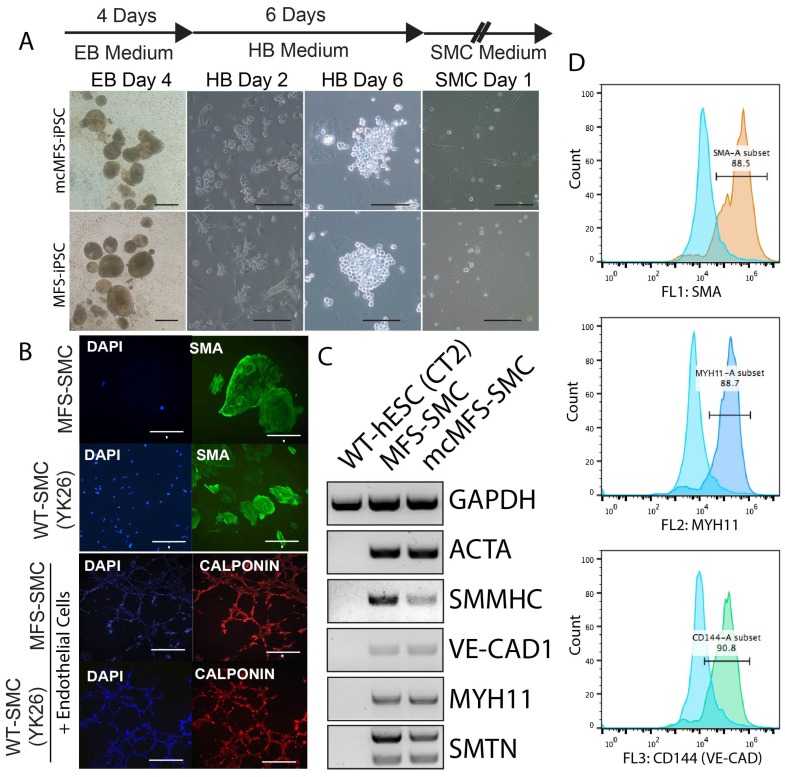
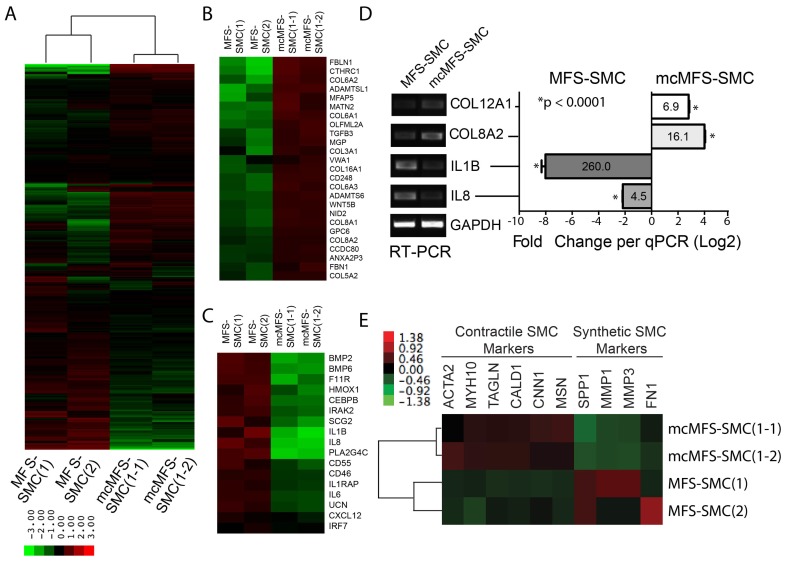
Marfan syndrome (MFS) is a connective tissue disorder caused by mutations in gene, which encodes a key extracellular matrix protein FIBRILLIN-1. The haplosufficiency of FBN1 has been implicated in pathogenesis of MFS with manifestations primarily in cardiovascular, muscular, and ocular tissues. Due to limitations in animal models to study the late-onset diseases, human pluripotent stem cells (PSCs) offer a homogeneic tool for dissection of cellular and molecular pathogenic mechanism for MFS . Here, we first derived induced PSCs (iPSCs) from a MFS patient with a mutation and corrected the mutation, thereby generating an isogenic "gain-of-function" control cells for the parental MFS iPSCs. Reversely, we knocked out in both alleles in a wild-type (WT) human embryonic stem cell (ESC) line, which served as a loss-of-function model for MFS with the WT cells as an isogenic control. Mesenchymal stem cells derived from both -mutant iPSCs and -ESCs demonstrated reduced osteogenic differentiation and microfibril formation. We further demonstrated that vascular smooth muscle cells derived from -mutant iPSCs showed less sensitivity to carbachol as demonstrated by contractility and Ca influx assay, compared to the isogenic controls cells. These findings were further supported by transcriptomic anaylsis of the cells. Therefore, this study based on both gain- and loss-of-function approaches confirmed the pathogenetic role of mutations in these MFS-related phenotypic changes.
马凡综合征(MFS)是一种由基因 突变引起的结缔组织疾病,该基因编码一种关键的细胞外基质蛋白原纤蛋白-1。FBN1的单倍剂量不足与MFS的发病机制有关,其表现主要在心血管、肌肉和眼部组织。由于动物模型在研究迟发性疾病方面存在局限性,人类多能干细胞(PSC)为剖析MFS的细胞和分子致病机制提供了一种同源工具。在这里,我们首先从一名携带 突变的MFS患者中获得诱导多能干细胞(iPSC)并纠正了突变,从而为亲代MFS iPSC生成了一个同基因的“功能获得”对照细胞。相反,我们在一个野生型(WT)人类胚胎干细胞(ESC)系的两个等位基因中敲除了 ,该细胞系作为MFS的功能丧失模型,以WT细胞作为同基因对照。来自 突变iPSC和 ESC的间充质干细胞均表现出成骨分化和微原纤维形成减少。我们进一步证明,与同基因对照细胞相比,通过收缩性和钙内流测定,来自 突变iPSC的血管平滑肌细胞对卡巴胆碱的敏感性较低。这些发现得到了细胞转录组分析的进一步支持。因此,这项基于功能获得和功能丧失方法的研究证实了 突变在这些与MFS相关的表型变化中的致病作用。