Toral Marcus A, Velez Gabriel, Boudreault Katherine, Schaefer Kellie A, Xu Yu, Saffra Norman, Bassuk Alexander G, Tsang Stephen H, Mahajan Vinit B
Omics LaboratoryUniversity of IowaIowa CityIowa.
Department of Ophthalmology and Visual SciencesUniversity of IowaIowa CityIowa.
Mol Genet Genomic Med. 2017 Feb 26;5(3):202-209. doi: 10.1002/mgg3.266. eCollection 2017 May.
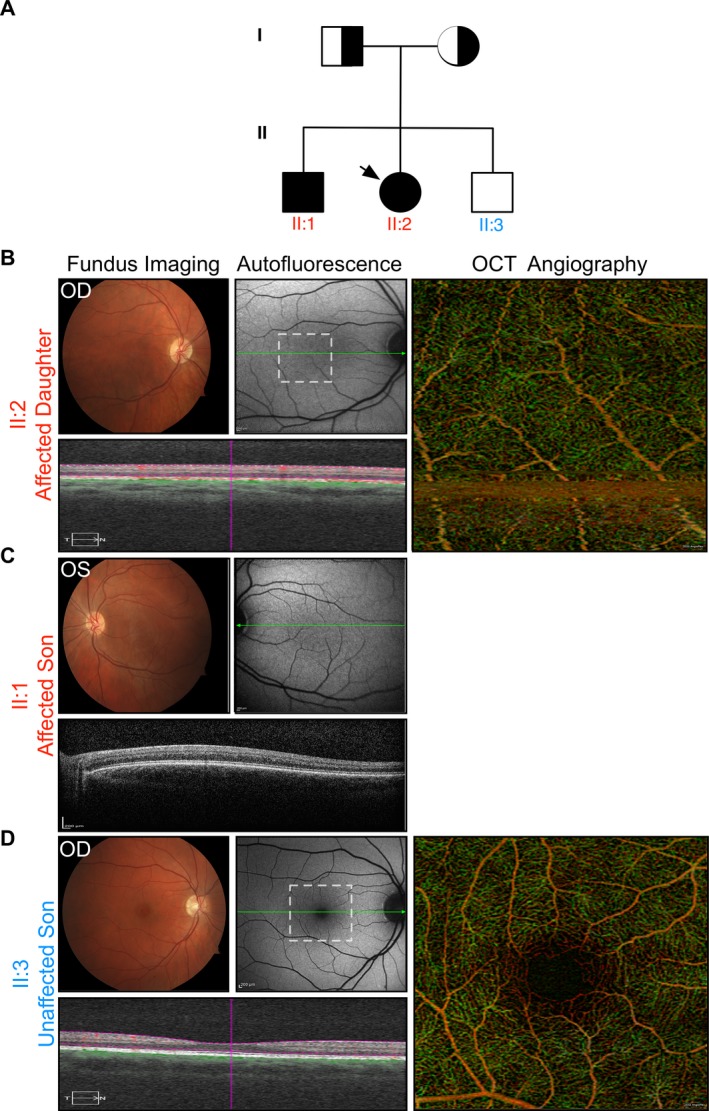
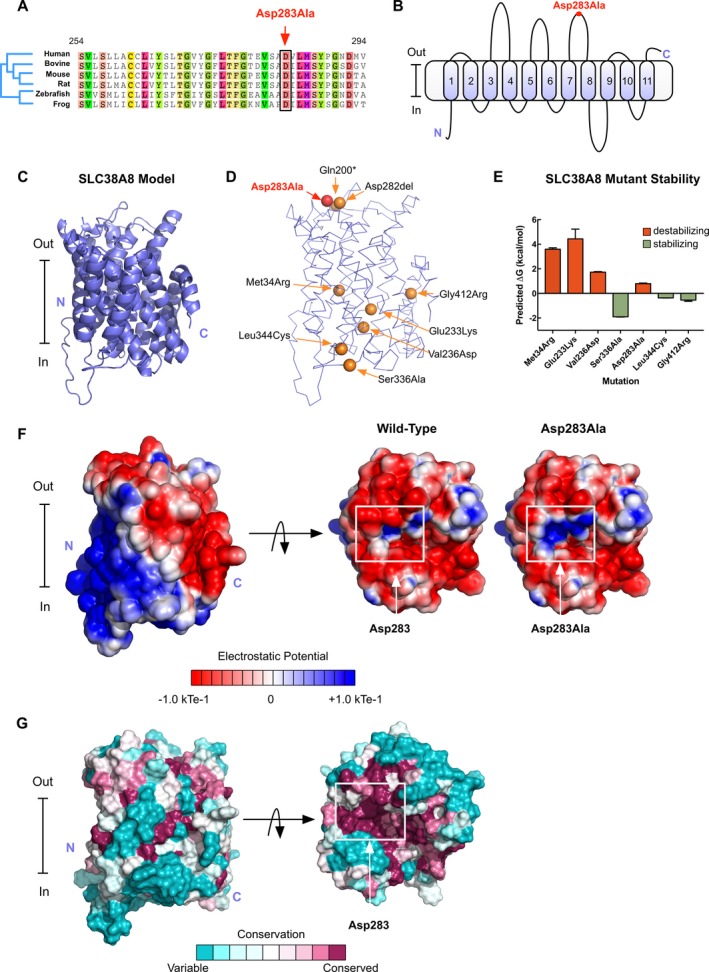
Foveal hypoplasia (FH) in the absence of albinism, aniridia, microphthalmia, or achromatopsia is exceedingly rare, and the molecular basis for the disorder remains unknown. FH is characterized by the absence of both the retinal foveal pit and avascular zone, but with preserved retinal architecture. encodes a sodium-coupled neutral amino acid transporter with a preference for glutamate as a substrate. SLC38A8 has been linked to FH. Here, we describe a novel mutation to which causes FH, and report the novel use of OCT-angiography to improve the precision of FH diagnosis. More so, we used computational modeling to explore possible functional effects of known SLC38A8 mutations.
Fundus autofluorescence, SD-OCT, and OCT-angiography were used to make the clinical diagnosis. Whole-exome sequencing led to the identification of a novel disease-causing variant in . Computational modeling approaches were used to visualize known SLC38A8 mutations, as well as to predict mutation effects on transporter structure and function.
We identified a novel point mutation in that causes FH. A conclusive diagnosis was made using OCT-angiography, which more clearly revealed retinal vasculature penetrating into the foveal region. Structural modeling of the channel showed the mutation was near previously published mutations, clustered on an extracellular loop. Our modeling also predicted that the mutation destabilizes the protein by altering the electrostatic potential within the channel pore.
Our results demonstrate a novel use for OCT-angiography in confirming FH, and also uncover genotype-phenotype correlations of FH-linked mutations.
在无白化病、无虹膜、小眼症或色盲的情况下,黄斑发育不全(FH)极为罕见,该病症的分子基础仍不清楚。FH的特征是视网膜黄斑中心凹凹陷和无血管区均缺失,但视网膜结构保留。SLC38A8编码一种偏好以谷氨酸为底物的钠偶联中性氨基酸转运体。SLC38A8已与FH相关联。在此,我们描述了一个导致FH的SLC38A8新突变,并报告了光学相干断层扫描血管造影术(OCT-angiography)在提高FH诊断准确性方面的新用途。此外,我们使用计算模型来探索已知SLC38A8突变可能的功能影响。
使用眼底自发荧光、谱域光学相干断层扫描(SD-OCT)和OCT-血管造影术进行临床诊断。全外显子组测序导致在SLC38A8中鉴定出一种新的致病变异。使用计算建模方法来可视化已知的SLC38A8突变,以及预测突变对转运体结构和功能的影响。
我们在SLC38A8中鉴定出一个导致FH的新点突变。使用OCT-血管造影术做出了确定性诊断,其更清晰地显示了视网膜血管穿透进入黄斑区域。该通道的结构建模显示该突变靠近先前发表的突变,聚集在一个细胞外环上。我们的建模还预测该突变通过改变通道孔内的静电势使蛋白质不稳定。
我们的结果证明了OCT-血管造影术在确认FH方面的新用途,并且还揭示了与FH相关的SLC38A8突变的基因型-表型相关性。