Narise Takafumi, Sakurai Nozomu, Obayashi Takeshi, Ohta Hiroyuki, Shibata Daisuke
Kazusa DNA Research Institute, 2-6-7 Kazusa-Kamatari, Kisarazu, Chiba, 292-0818, Japan.
Graduate School of Information Sciences, Tohoku University, 6-3-09 Aramaki-Aza-Aoba, Aoba-ku, Sendai, Miyagi, 980-8579, Japan.
BMC Genomics. 2017 Jun 5;18(1):437. doi: 10.1186/s12864-017-3786-3.
Gene co-expression, the similarity of gene expression profiles under various experimental conditions, has been used as an indicator of functional relationships between genes, and many co-expression databases have been developed for predicting gene functions. These databases usually provide users with a co-expression network and a list of strongly co-expressed genes for a query gene. Several of these databases also provide functional information on a set of strongly co-expressed genes (i.e., provide biological processes and pathways that are enriched in these strongly co-expressed genes), which is generally analyzed via over-representation analysis (ORA). A limitation of this approach may be that users can predict gene functions only based on the strongly co-expressed genes.
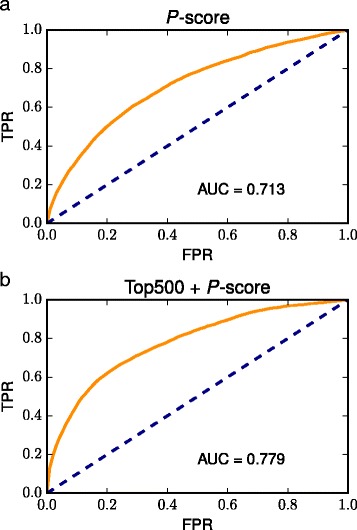
In this study, we developed a new co-expression database that enables users to predict the function of tomato genes from the results of functional enrichment analyses of co-expressed genes while considering the genes that are not strongly co-expressed. To achieve this, we used the ORA approach with several thresholds to select co-expressed genes, and performed gene set enrichment analysis (GSEA) applied to a ranked list of genes ordered by the co-expression degree. We found that internal correlation in pathways affected the significance levels of the enrichment analyses. Therefore, we introduced a new measure for evaluating the relationship between the gene and pathway, termed the percentile (p)-score, which enables users to predict functionally relevant pathways without being affected by the internal correlation in pathways. In addition, we evaluated our approaches using receiver operating characteristic curves, which concluded that the p-score could improve the performance of the ORA.
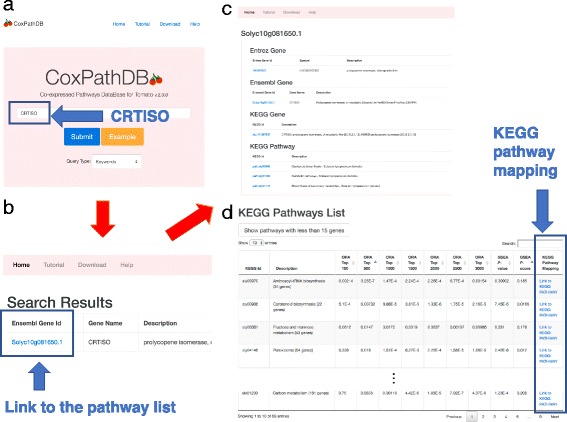
We developed a new database, named Co-expressed Pathways DataBase for Tomato, which is available at http://cox-path-db.kazusa.or.jp/tomato . The database allows users to predict pathways that are relevant to a query gene, which would help to infer gene functions.
基因共表达,即在各种实验条件下基因表达谱的相似性,已被用作基因间功能关系的指标,并且已经开发了许多共表达数据库来预测基因功能。这些数据库通常为用户提供一个共表达网络以及一个查询基因的强共表达基因列表。其中一些数据库还提供一组强共表达基因的功能信息(即提供在这些强共表达基因中富集的生物学过程和途径),这些信息通常通过超几何富集分析(ORA)进行分析。这种方法的一个局限性可能是用户只能基于强共表达基因来预测基因功能。
在本研究中,我们开发了一个新的共表达数据库,该数据库使用户能够在考虑非强共表达基因的情况下,根据共表达基因的功能富集分析结果预测番茄基因的功能。为了实现这一目标,我们使用具有多个阈值的ORA方法来选择共表达基因,并对按共表达程度排序的基因列表进行基因集富集分析(GSEA)。我们发现途径中的内部相关性会影响富集分析的显著性水平。因此,我们引入了一种评估基因与途径之间关系的新方法,称为百分位数(p)得分,它使用户能够预测功能相关途径而不受途径内部相关性的影响。此外,我们使用受试者工作特征曲线评估了我们的方法,结果表明p得分可以提高ORA的性能。
我们开发了一个名为番茄共表达途径数据库(Co-expressed Pathways DataBase for Tomato)的新数据库,可通过http://cox-path-db.kazusa.or.jp/tomato访问。该数据库允许用户预测与查询基因相关的途径,这将有助于推断基因功能。