Division of Genetics and Genomics, Manton Center for Orphan Disease, and Howard Hughes Medical Institute, Boston Children's Hospital, Boston, Massachusetts 02115, USA.
Departments of Neurology and Pediatrics, Harvard Medical School, Boston, Massachusetts 02115, USA.
Genome Res. 2017 Aug;27(8):1323-1335. doi: 10.1101/gr.219899.116. Epub 2017 Jun 19.
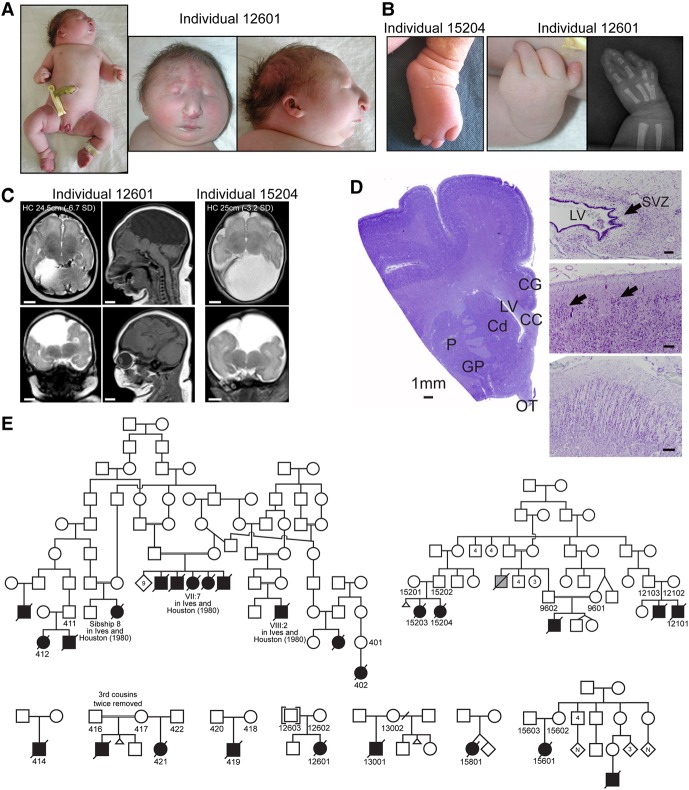
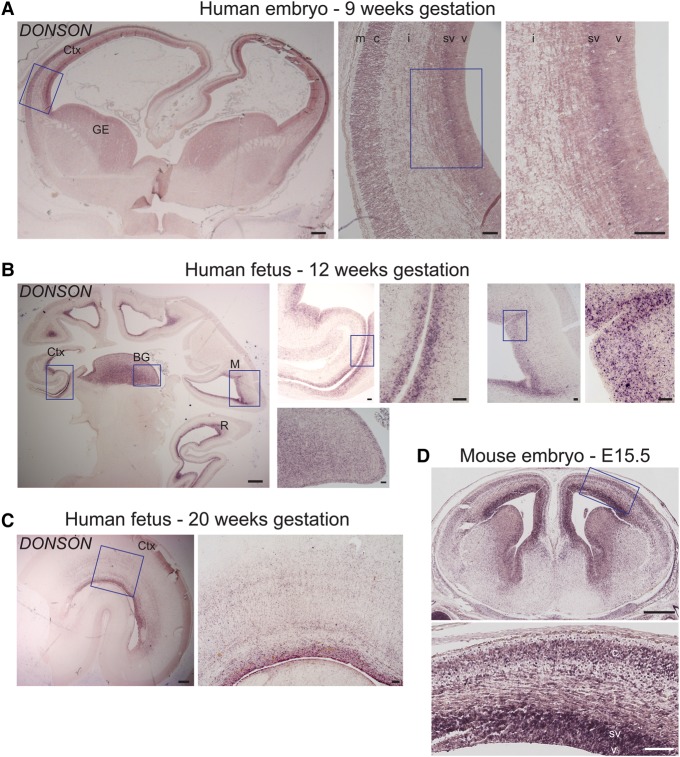
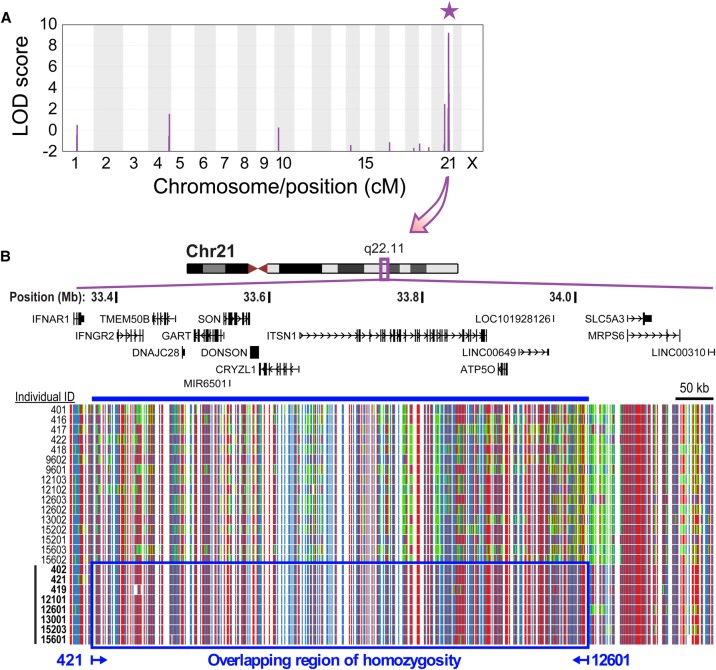
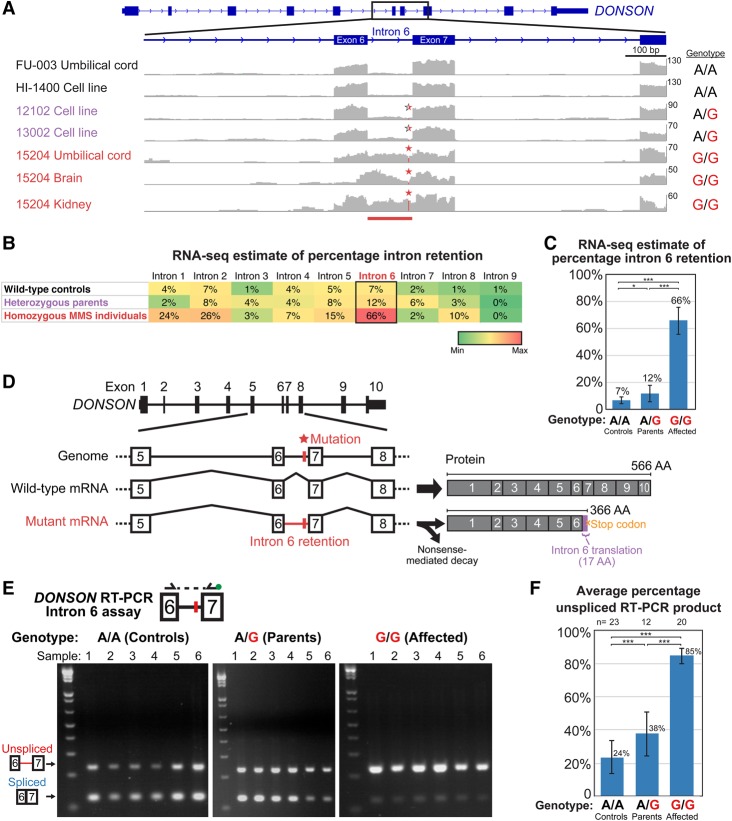
While next-generation sequencing has accelerated the discovery of human disease genes, progress has been largely limited to the "low hanging fruit" of mutations with obvious exonic coding or canonical splice site impact. In contrast, the lack of high-throughput, unbiased approaches for functional assessment of most noncoding variants has bottlenecked gene discovery. We report the integration of transcriptome sequencing (RNA-seq), which surveys all mRNAs to reveal functional impacts of variants at the transcription level, into the gene discovery framework for a unique human disease, microcephaly-micromelia syndrome (MMS). MMS is an autosomal recessive condition described thus far in only a single First Nations population and causes intrauterine growth restriction, severe microcephaly, craniofacial anomalies, skeletal dysplasia, and neonatal lethality. Linkage analysis of affected families, including a very large pedigree, identified a single locus on Chromosome 21 linked to the disease (LOD > 9). Comprehensive genome sequencing did not reveal any pathogenic coding or canonical splicing mutations within the linkage region but identified several nonconserved noncoding variants. RNA-seq analysis detected aberrant splicing in due to one of these noncoding variants, showing a causative role for disruption in MMS. We show that is expressed in progenitor cells of embryonic human brain and other proliferating tissues, is co-expressed with components of the DNA replication machinery, and that is essential for early embryonic development in mice as well, suggesting an essential conserved role for DONSON in the cell cycle. Our results demonstrate the utility of integrating transcriptomics into the study of human genetic disease when DNA sequencing alone is not sufficient to reveal the underlying pathogenic mutation.
虽然下一代测序技术加速了人类疾病基因的发现,但进展主要局限于具有明显外显子编码或规范剪接位点影响的“低垂果实”突变。相比之下,缺乏高通量、无偏的方法来对大多数非编码变体进行功能评估,这限制了基因的发现。我们报告了转录组测序(RNA-seq)的整合,该技术可以全面检测所有 mRNA,从而在转录水平上揭示变体的功能影响,并将其整合到一个独特的人类疾病——小头-短肢综合征(MMS)的基因发现框架中。MMS 是一种常染色体隐性疾病,迄今为止仅在一个第一民族群体中描述过,它会导致宫内生长受限、严重的小头畸形、颅面异常、骨骼发育不良和新生儿死亡。受影响的家族(包括一个非常大的家系)的连锁分析确定了 21 号染色体上与该疾病相关的一个单一基因座(LOD>9)。全基因组测序并未在连锁区域内发现任何致病性编码或规范剪接突变,但鉴定出了几个非保守的非编码变体。RNA-seq 分析检测到由于其中一个非编码变体导致 发生异常剪接,表明 破坏在 MMS 中起因果作用。我们表明 表达于胚胎人脑和其他增殖组织的祖细胞中,与 DNA 复制机制的组件共同表达,并且 在小鼠中对早期胚胎发育也是必需的,这表明 DONSON 在细胞周期中具有保守的重要作用。我们的结果表明,当单独进行 DNA 测序不足以揭示潜在的致病突变时,将转录组学整合到人类遗传疾病研究中是有用的。