Popova Evgenya Y, Salzberg Anna C, Yang Chen, Zhang Samuel Shao-Min, Barnstable Colin J
Department of Neural and Behavioral Sciences, Penn State University, College of Medicine, Hershey, Pennsylvania, United States of America.
Penn State Hershey Eye Center, Hershey, Pennsylvania, United States of America.
PLoS One. 2017 Jun 22;12(6):e0179230. doi: 10.1371/journal.pone.0179230. eCollection 2017.
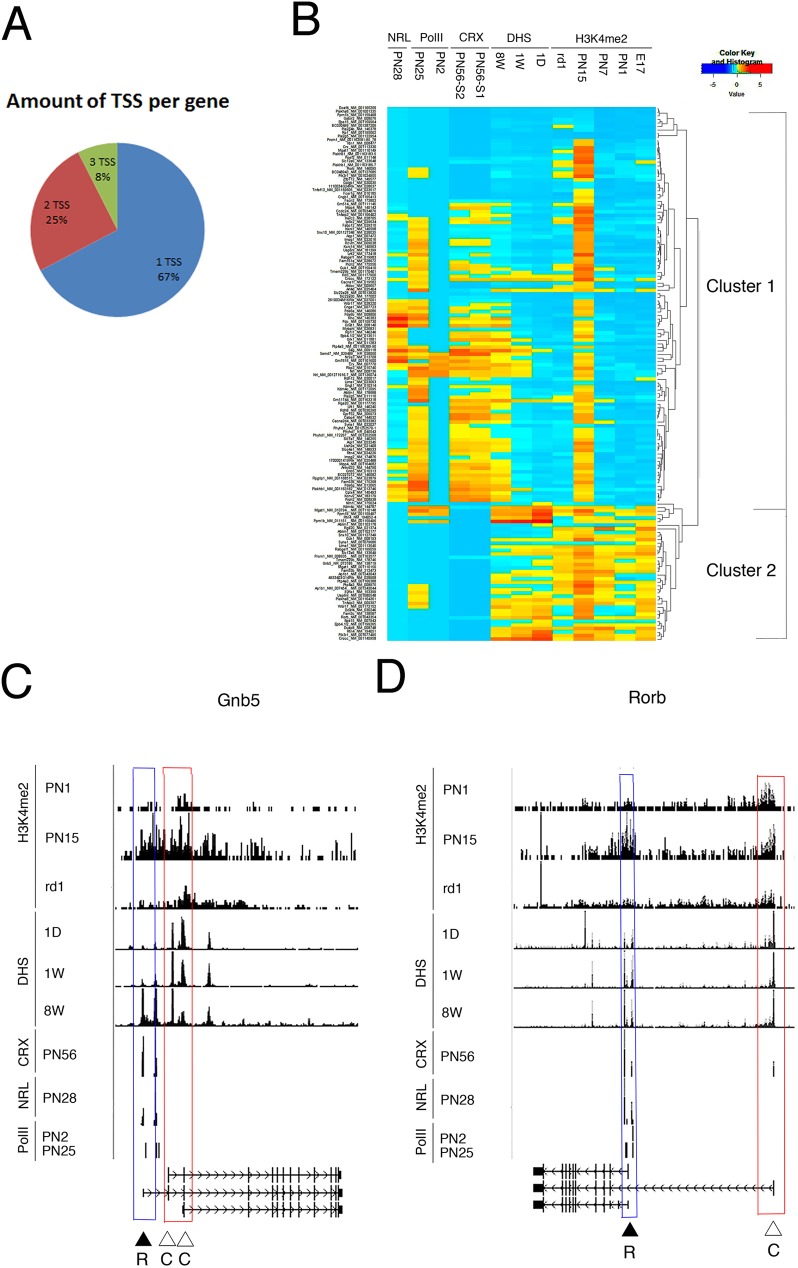
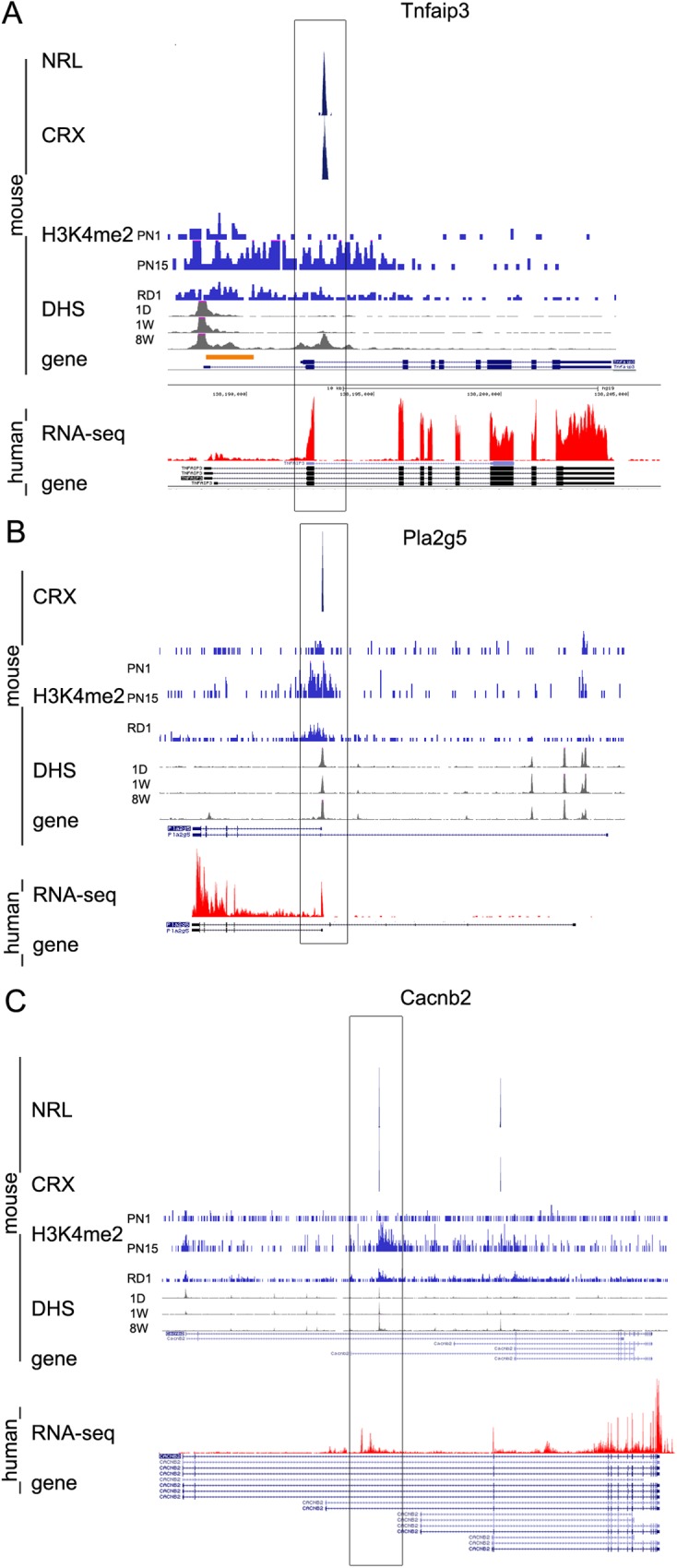
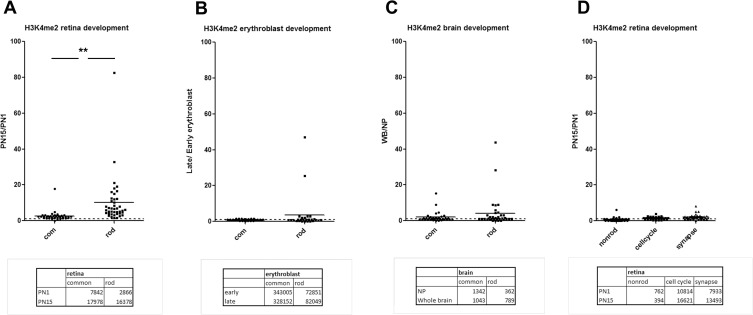
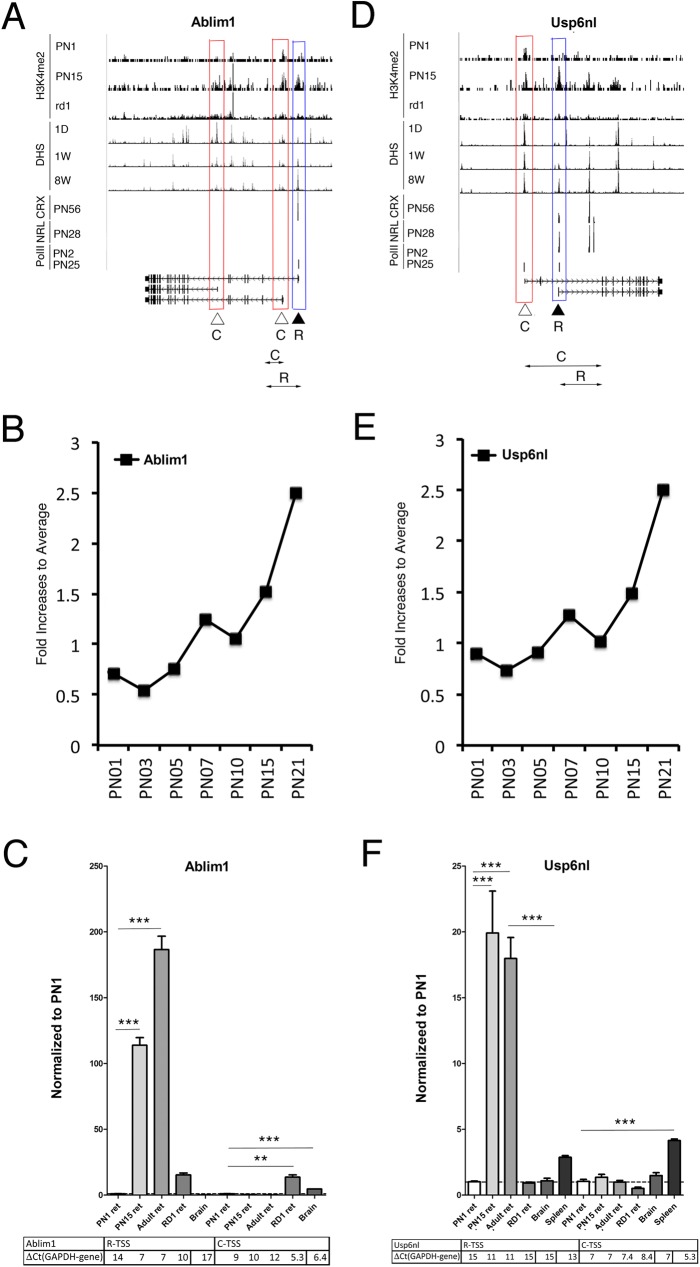
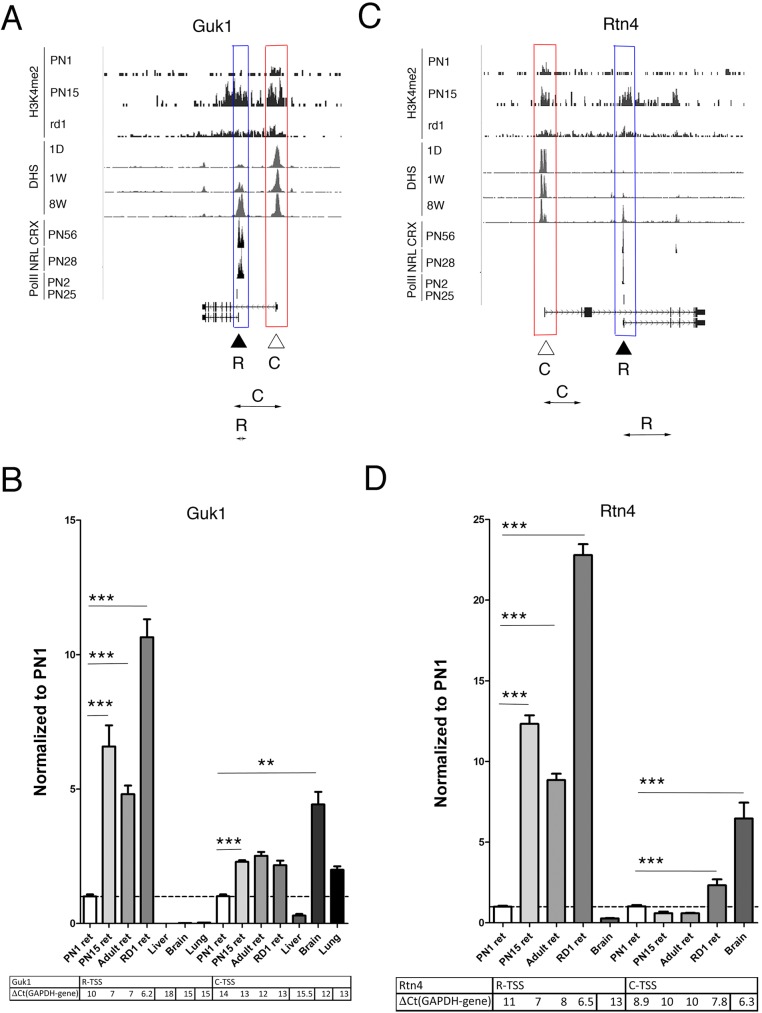
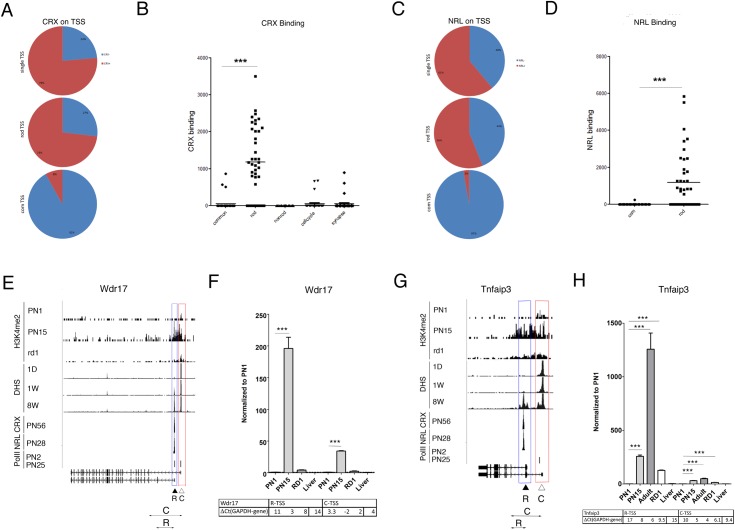
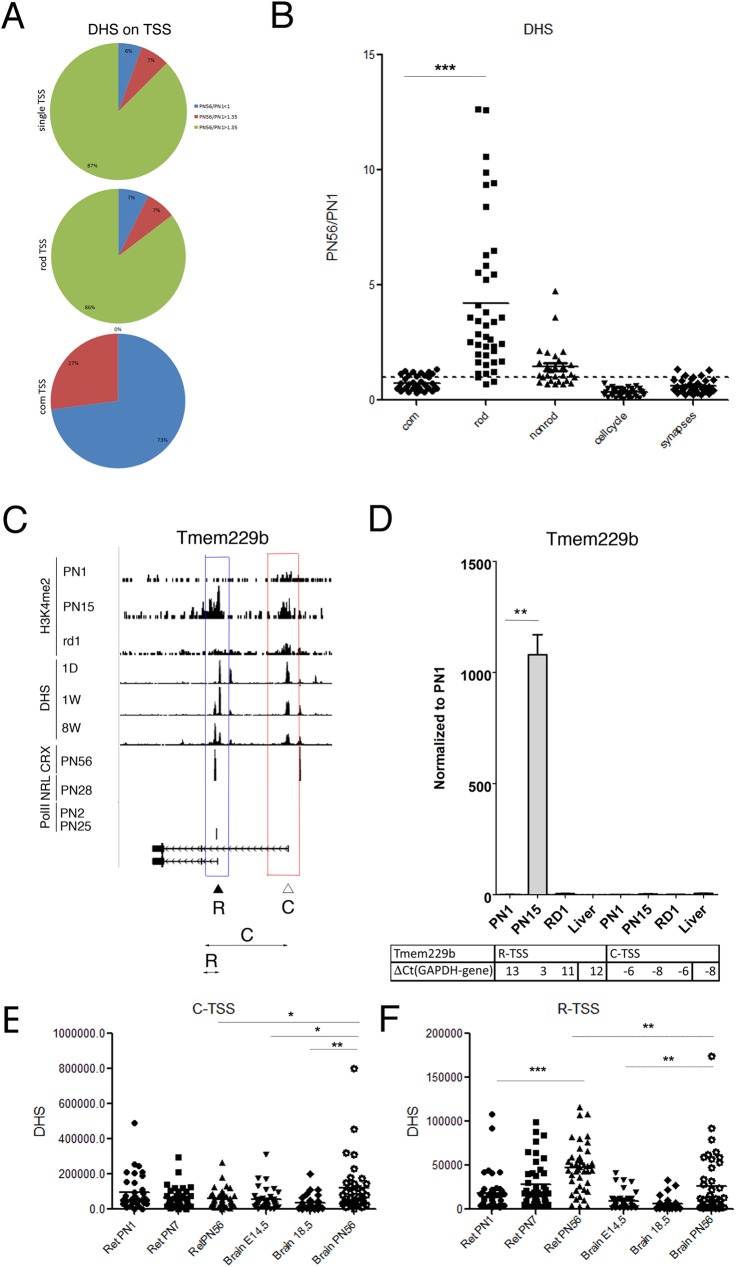
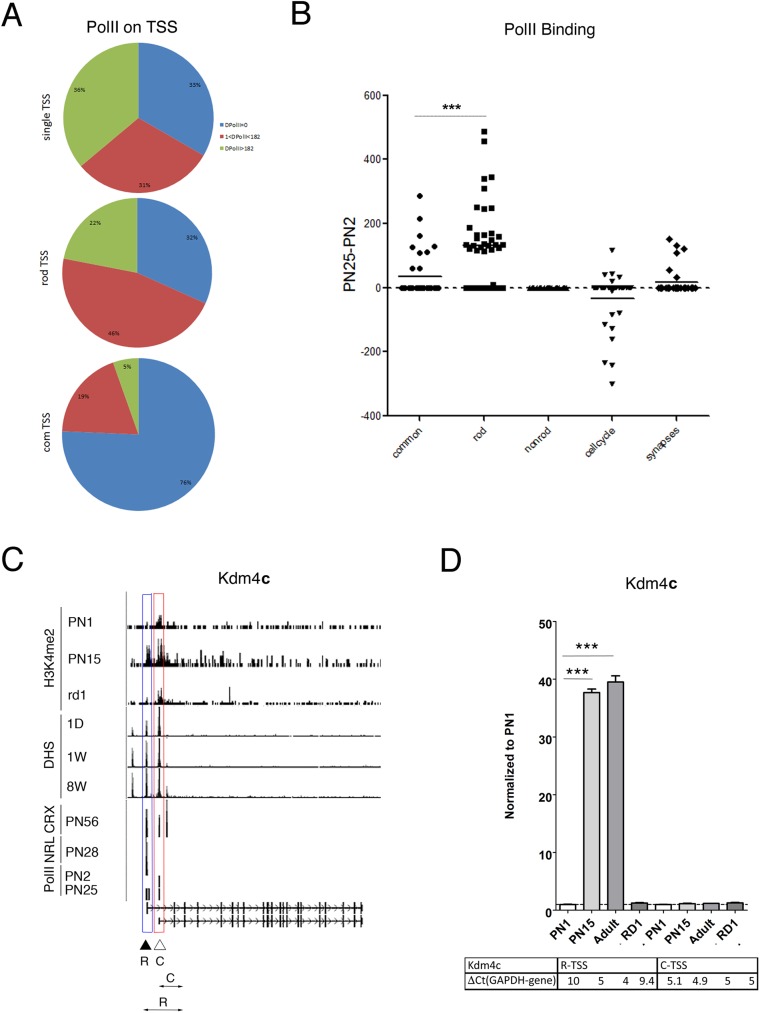
Transcriptome complexity is substantially increased by the use of multiple transcription start sites for a given gene. By utilizing a rod photoreceptor-specific chromatin signature, and the RefSeq database of established transcription start sites, we have identified essentially all known rod photoreceptor genes as well as a group of novel genes that have a high probability of being expressed in rod photoreceptors. Approximately half of these novel rod genes are transcribed into multiple mRNA and/or protein isoforms through alternative transcriptional start sites (ATSS), only one of which has a rod-specific epigenetic signature and gives rise to a rod transcript. This suggests that, during retina development, some genes use ATSS to regulate cell type and temporal specificity, effectively generating a rod transcript from otherwise ubiquitously expressed genes. Biological confirmation of the relationship between epigenetic signatures and gene expression, as well as comparison of our genome-wide chromatin signature maps with available data sets for retina, namely a ChIP-on-Chip study of Polymerase-II (Pol-II) binding sites, ChIP-Seq studies for NRL- and CRX- binding sites and DHS (University of Washington data, available on UCSC mouse Genome Browser as a part of ENCODE project) fully support our hypothesis and together accurately identify and predict an array of new rod transcripts. The same approach was used to identify a number of TSS that are not currently in RefSeq. Biological conformation of the use of some of these TSS suggests that this method will be valuable for exploring the range of transcriptional complexity in many tissues. Comparison of mouse and human genome-wide data indicates that most of these alternate TSS appear to be present in both species, indicating that our approach can be useful for identification of regulatory regions that might play a role in human retinal disease.
对于给定基因,使用多个转录起始位点可显著增加转录组的复杂性。通过利用视杆光感受器特异性染色质特征以及已建立的转录起始位点的RefSeq数据库,我们已确定了基本上所有已知的视杆光感受器基因以及一组极有可能在视杆光感受器中表达的新基因。这些新的视杆基因中约有一半通过可变转录起始位点(ATSS)转录成多种mRNA和/或蛋白质异构体,其中只有一种具有视杆特异性表观遗传特征并产生视杆转录本。这表明,在视网膜发育过程中,一些基因利用ATSS来调节细胞类型和时间特异性,有效地从原本广泛表达的基因中产生视杆转录本。表观遗传特征与基因表达之间关系的生物学验证,以及我们全基因组染色质特征图谱与视网膜现有数据集(即聚合酶II(Pol-II)结合位点的芯片杂交研究、NRL和CRX结合位点以及DHS的ChIP-Seq研究(华盛顿大学数据,作为ENCODE项目的一部分可在UCSC小鼠基因组浏览器上获取))的比较,充分支持了我们的假设,并共同准确地识别和预测了一系列新的视杆转录本。同样的方法被用于识别一些目前不在RefSeq中的转录起始位点(TSS)。对其中一些TSS使用情况的生物学验证表明,该方法对于探索许多组织中的转录复杂性范围将是有价值的。小鼠和人类全基因组数据的比较表明,这些可变TSS中的大多数似乎在两个物种中都存在,这表明我们的方法可用于识别可能在人类视网膜疾病中起作用的调控区域。