Kim Katherine H, Messinger Yoav H, Burton Barbara K
Ann & Robert H. Lurie Children's Hospital of Chicago, Northwestern University, Feinberg School of Medicine, United States.
Children's Hospitals and Clinics of Minnesota, United States.
Mol Genet Metab Rep. 2014 Nov 27;2:20-24. doi: 10.1016/j.ymgmr.2014.11.007. eCollection 2015 Mar.
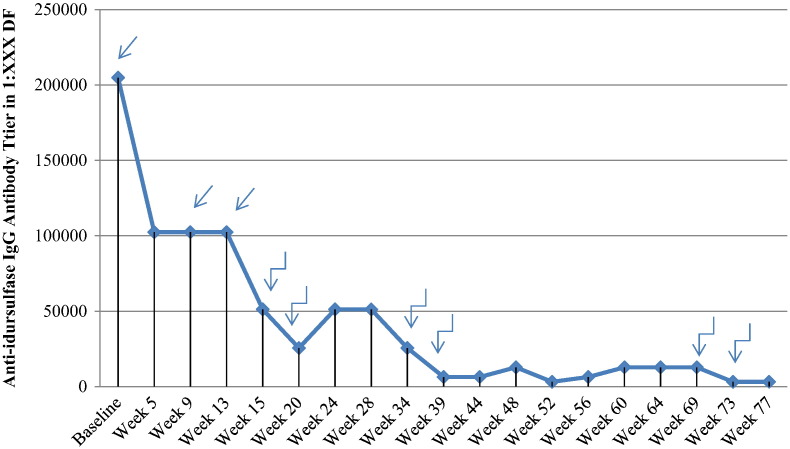
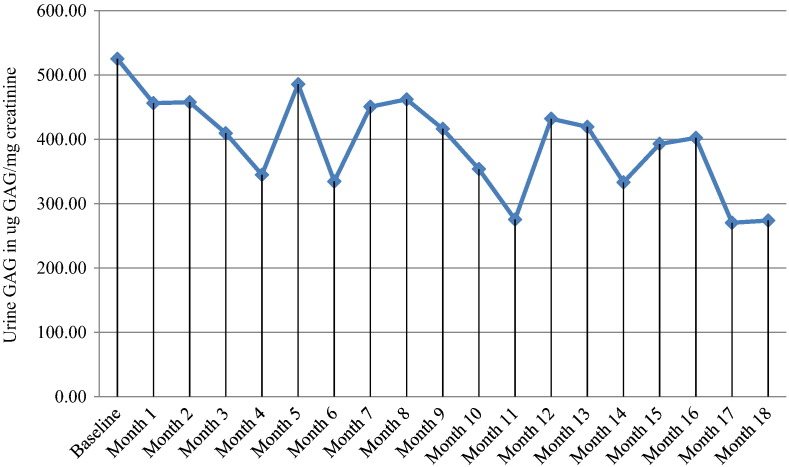
We report on a 6 year old boy with severe MPS II undergoing immune modulation therapy due to high IgG antibody titers to IV idursulfase and no significant decline in urinary GAG levels since initiating enzyme replacement therapy. He has complete deficiency of iduronate-2-sulfatase activity due to a submicroscopic deletion of the X chromosome involving the entire I2S gene but not including in the fragile X locus. At 19 months of age, IV idursulfase therapy at the recommended dose of 0.5 mg/kg/week was initiated and then increased to 1.0 mg/kg/week after no observed clinical improvement and no decline in urine GAG level. After one year of ERT at the increased dose, he had no significant decline in urinary GAG excretion and increase of anti-idursulfase IgG antibody titers to 102,000 with complete neutralizing antibodies. In light of the evidence of lack of efficacy of idursulfase therapy, the patient was started on an immune modulation regimen consisting of ofatumumab, bortezomib, methotrexate and IVIG for a 12 week period. Only a slight decrease in IgG titers and urine GAG levels was observed, leading to increased intensity of bortezomib administration and addition of dexamethasone to the regimen, while continuing with the current schedule ofatumumab, IVIG and methotrexate. Over 18 month period of immune modulation therapy, we observed a significant reduction in anti-idursulfase IgG titers and a moderate reduction in urine GAG levels compared to baseline. Modest clinical improvements were observed. Our experience suggests that future MPS II patients with a complete gene deletion may be likely to develop persistent anti-idursulfase antibody titers and may benefit from immune modulation therapy prior to the development of high titer levels.
我们报告了一名6岁患有严重II型黏多糖贮积症(MPS II)的男孩,由于其对静脉注射艾度硫酸酯酶的IgG抗体滴度很高,且自开始酶替代疗法以来尿糖胺聚糖(GAG)水平没有显著下降,正在接受免疫调节治疗。由于X染色体发生亚显微缺失,累及整个艾杜糖醛酸-2-硫酸酯酶(I2S)基因但不包括脆性X位点,他完全缺乏艾杜糖醛酸-2-硫酸酯酶活性。19个月大时,开始以推荐剂量0.5mg/kg/周进行静脉注射艾度硫酸酯酶治疗,在未观察到临床改善且尿GAG水平未下降后,剂量增加至1.0mg/kg/周。在增加剂量的酶替代疗法(ERT)治疗一年后,他的尿GAG排泄量没有显著下降,抗艾度硫酸酯酶IgG抗体滴度增加到102,000,且产生了完全中和抗体。鉴于艾度硫酸酯酶治疗缺乏疗效的证据,该患者开始接受为期12周的免疫调节方案,包括奥法木单抗、硼替佐米、甲氨蝶呤和静脉注射免疫球蛋白(IVIG)。仅观察到IgG滴度和尿GAG水平略有下降,导致硼替佐米给药强度增加,并在方案中添加地塞米松,同时继续当前的奥法木单抗、IVIG和甲氨蝶呤给药方案。在为期18个月的免疫调节治疗期间,与基线相比,我们观察到抗艾度硫酸酯酶IgG滴度显著降低,尿GAG水平适度降低。观察到了适度的临床改善。我们的经验表明,未来患有完全基因缺失的MPS II患者可能会产生持续的抗艾度硫酸酯酶抗体滴度,并且在高滴度水平出现之前可能会从免疫调节治疗中获益。