Sperb-Ludwig F, Alegra T, Velho R V, Ludwig N, Kim C A, Kok F, Kitajima J P, van Meel E, Kornfeld S, Burin M G, Schwartz I V D
BRAIN (Basic Research and Advanced Investigations in Neurosciences) Laboratory, Hospital de Clínicas de Porto Alegre (HCPA), Brazil.
Postgraduate Program in Medical Sciences: Medical Sciences, Universidade Federal do Rio Grande do Sul (UFRGS), Brazil.
Mol Genet Metab Rep. 2014 Dec 5;2:34-37. doi: 10.1016/j.ymgmr.2014.12.001. eCollection 2015 Mar.
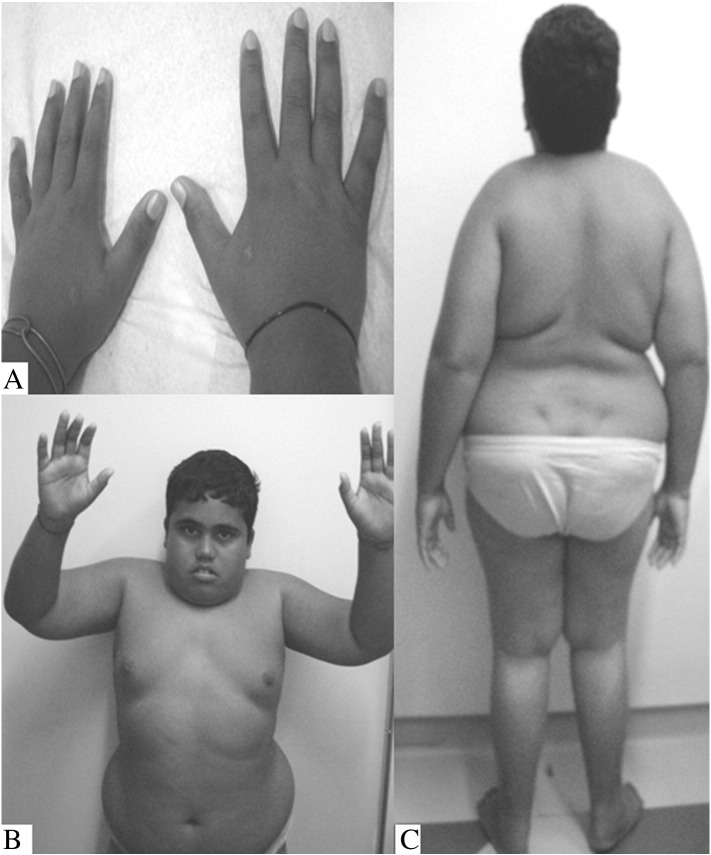
Mucolipidosis II and III alpha/beta (ML II/III alpha/beta) are rare autosomal recessive lysosomal storage diseases that are caused by a deficiency of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase, the enzyme responsible for the synthesis of the mannose 6-phosphate targeting signal on lysosomal hydrolases. A Brazilian patient suspected of having a very mild ML III was investigated using whole next-generation sequencing (NGS). Two mutations in the gene were detected and confirmed to be status by parental analysis: c.1208T>C (p.Ile403Thr), previously reported as being pathogenic, and the novel mutation c.1723G>A (p.Gly575Arg). This study demonstrates the effectiveness of using whole NGS for the molecular diagnosis of very mild ML III alpha/beta patients.
粘脂贮积症II型和III型α/β亚型(ML II/III α/β)是罕见的常染色体隐性溶酶体贮积病,由UDP-GlcNAc:溶酶体酶N-乙酰葡糖胺-1-磷酸转移酶缺乏所致,该酶负责在溶酶体水解酶上合成甘露糖6-磷酸靶向信号。一名疑似患有非常轻度ML III型的巴西患者通过全基因组二代测序(NGS)进行了研究。在该基因中检测到两个突变,并通过亲代分析确认为致病状态:c.1208T>C(p.Ile403Thr),此前报道为致病性突变,以及新突变c.1723G>A(p.Gly575Arg)。本研究证明了使用全基因组NGS对非常轻度的ML III α/β型患者进行分子诊断的有效性。