Fukushima Masakazu, Iizuka Kenzo, Jin Cheng, Zhang Chun, Hong Mei, Eshima Kiyoshi
Division of Oncology Research and Development, Delta-Fly Pharma Inc., Kawauchi-cho, Tokushima, Japan.
Drug Des Devel Ther. 2017 Jun 7;11:1693-1705. doi: 10.2147/DDDT.S128420. eCollection 2017.
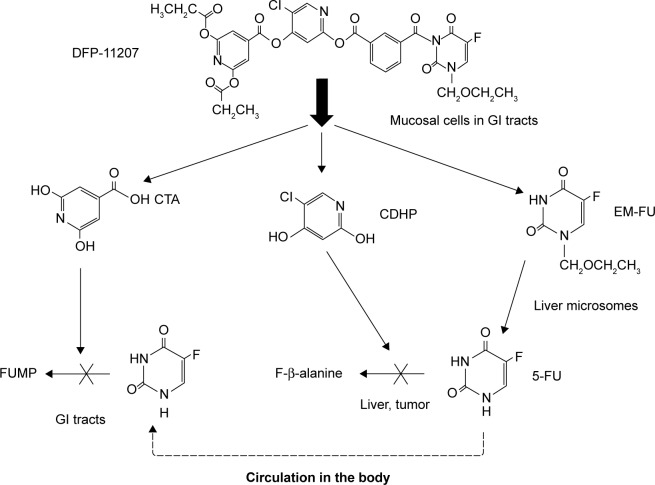
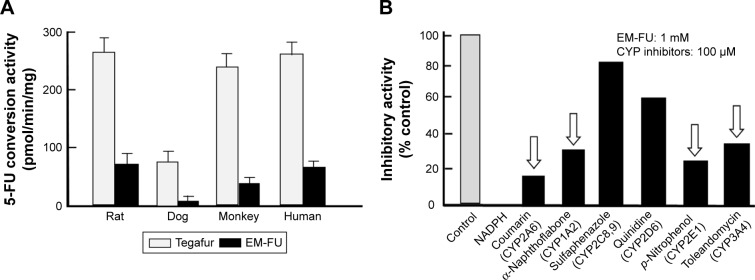
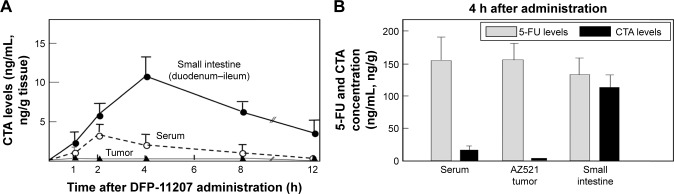
To reduce 5-fluorouracil (5-FU)-induced serious toxicities without loss of antitumor activity, we have developed DFP-11207, a novel fluoropyrimidine, which consists of 1-ethoxymethyl-5-fluorouracil (EM-FU; a precursor form of 5-FU), 5-chloro-2,4-dihydroxypyridine (CDHP; an inhibitor of 5-FU degradation), and citrazinic acid (CTA; an inhibitor of 5-FU phosphorylation). In vitro studies of DFP-11207 indicated that it strongly inhibited the degradation of 5-FU by dihydropyrimidine dehydrogenase (DPD) in homogenates of the rat liver, and also inhibited the phosphorylation of 5-FU by orotate phosphoribosyltransferase (OPRT) in tumor tissues in a similar magnitude of potency by CDHP and CTA, respectively. Especially, DFP-11207 inhibited the intracellular phosphorylation of 5-FU in tumor cells in a dose-dependent manner whereas CTA alone did not protect intracellular 5-FU phosphorylation. These results postulate that DFP-11207 rapidly entered into the cell and the free CTA produced from DFP-11207 inhibited the phosphorylation of 5-FU in the cell. Furthermore, following oral administration of DFP-11207, CTA was found to be highly retained in the gastrointestinal (GI) tract compared to other tissues in rats. Interestingly, EM-FU, the prodrug of 5-FU was found to specifically produce 5-FU by various species of liver microsomes. When DFP-11207 was administered to rats, the plasma level of 5-FU was persisted for a long-time with lower C and longer half-life than that from other 5-FU prodrugs. The antitumor activity of DFP-11207 was evaluated in human tumor xenografts in nude rats and found that DFP-11207 showed an antitumor activity in a dose-dependent fashion and its efficacy is equivalent to reference 5-FU drugs. In striking contrast, DFP-11207 manifested no or less 5-FU-related toxicities, such as a decrease in body weights, GI injury, and myelosuppression, especially thrombocytopenia. Taken together, the preclinical evaluation of DFP-11207 strongly indicates that DFP-11207 be a potential new version of the oral fluoropyrimidine prodrug for further clinical development.
为了降低5-氟尿嘧啶(5-FU)诱导的严重毒性而不丧失抗肿瘤活性,我们研发了DFP-11207,一种新型氟嘧啶,它由1-乙氧甲基-5-氟尿嘧啶(EM-FU;5-FU的前体形式)、5-氯-2,4-二羟基吡啶(CDHP;5-FU降解抑制剂)和柠嗪酸(CTA;5-FU磷酸化抑制剂)组成。DFP-11207的体外研究表明,它能强烈抑制大鼠肝脏匀浆中二氢嘧啶脱氢酶(DPD)对5-FU的降解,并且分别以与CDHP和CTA相似的效力抑制肿瘤组织中乳清酸磷酸核糖基转移酶(OPRT)对5-FU的磷酸化。特别地,DFP-11207以剂量依赖的方式抑制肿瘤细胞中5-FU的细胞内磷酸化,而单独的CTA并不能保护细胞内5-FU的磷酸化。这些结果推测DFP-11207迅速进入细胞,并且由DFP-11207产生的游离CTA抑制细胞内5-FU的磷酸化。此外,口服DFP-11207后,发现与大鼠的其他组织相比,CTA在胃肠道(GI)中高度保留。有趣的是,发现5-FU的前体药物EM-FU能被各种肝微粒体特异性地转化为5-FU。当给大鼠施用DFP-11207时,5-FU的血浆水平长时间持续存在,与其他5-FU前体药物相比,其血药浓度(C)较低但半衰期较长。在裸鼠的人肿瘤异种移植模型中评估了DFP-11207的抗肿瘤活性,发现DFP-11207以剂量依赖的方式显示出抗肿瘤活性,并且其疗效与参考5-FU药物相当。与之形成鲜明对比的是,DFP-11207表现出无或较少的与5-FU相关的毒性,如体重减轻、胃肠道损伤和骨髓抑制,尤其是血小板减少。综上所述,DFP-11207的临床前评估强烈表明DFP-11207是一种有潜力的新型口服氟嘧啶前体药物以供进一步的临床开发。