Yu Baoqi, Kiechl Stefan, Qi Dan, Wang Xiaocong, Song Yanting, Weger Siegfried, Mayr Agnes, Le Bras Alexandra, Karamariti Eirini, Zhang Zhongyi, Barco Barrantes Ivan Del, Niehrs Christof, Schett Georg, Hu Yanhua, Wang Wen, Willeit Johann, Qu Aijuan, Xu Qingbo
From Cardiovascular Division, King's College London British Heart Foundation Centre, London, United Kingdom (B.Y., X.W., A.L.B., E.K., Z.Z., Y.H., Q.X.); Department of Neurology, Medical University of Innsbruck, Austria (S.K., J.W.); Department of Physiology and Pathophysiology, Capital Medical University, Beijing, China (D.Q., Y.S., A.Q.); Department of Internal and Laboratory Medicine, Bruneck Hospital, Italy (S.W., A.M.); Division of Molecular Embryology, German Cancer Research Center (DKFZ) Heidelberg Germany and Zentrum für Molekulare Biologie der Universität Heidelberg (ZMBH) Alliance, Heidelberg, Germany (I.d.B.B., C.N.); Institute of Molecular Biology, Mainz, Germany (C.N.); Department of Internal Medicine, Institute for Clinical Immunology, Friedrich-Alexander-University Erlangen-Nuremberg, Germany (G.S.); The Key Laboratory of Cardiovascular Remodelling and Function Research, Chinese Ministry of Education and Chinese Ministry of Health, Qilu Hospital, Shandong University, Jinan, China (Y.H., Q.X.); and Institute of Bioengineering, Queen Mary University of London, United Kingdom (W.W.).
From Cardiovascular Division, King's College London British Heart Foundation Centre, London, United Kingdom (B.Y., X.W., A.L.B., E.K., Z.Z., Y.H., Q.X.); Department of Neurology, Medical University of Innsbruck, Austria (S.K., J.W.); Department of Physiology and Pathophysiology, Capital Medical University, Beijing, China (D.Q., Y.S., A.Q.); Department of Internal and Laboratory Medicine, Bruneck Hospital, Italy (S.W., A.M.); Division of Molecular Embryology, German Cancer Research Center (DKFZ) Heidelberg Germany and Zentrum für Molekulare Biologie der Universität Heidelberg (ZMBH) Alliance, Heidelberg, Germany (I.d.B.B., C.N.); Institute of Molecular Biology, Mainz, Germany (C.N.); Department of Internal Medicine, Institute for Clinical Immunology, Friedrich-Alexander-University Erlangen-Nuremberg, Germany (G.S.); The Key Laboratory of Cardiovascular Remodelling and Function Research, Chinese Ministry of Education and Chinese Ministry of Health, Qilu Hospital, Shandong University, Jinan, China (Y.H., Q.X.); and Institute of Bioengineering, Queen Mary University of London, United Kingdom (W.W.)
Circulation. 2017 Sep 12;136(11):1022-1036. doi: 10.1161/CIRCULATIONAHA.117.027690. Epub 2017 Jul 3.
Dickkopf-related protein 3 (DKK3) is a secreted protein that is involved in the regulation of cardiac remodeling and vascular smooth muscle cell differentiation, but little is known about its role in atherosclerosis.
We tested the hypothesis that DKK3 is atheroprotective using both epidemiological and experimental approaches. Blood DKK3 levels were measured in the Bruneck Study in 2000 (n=684) and then in 2005 (n=574). -deficient mice were crossed with mice to evaluate atherosclerosis development and vessel injury-induced neointimal formation. Endothelial cell migration and the underlying mechanisms were studied using in vitro cell culture models.
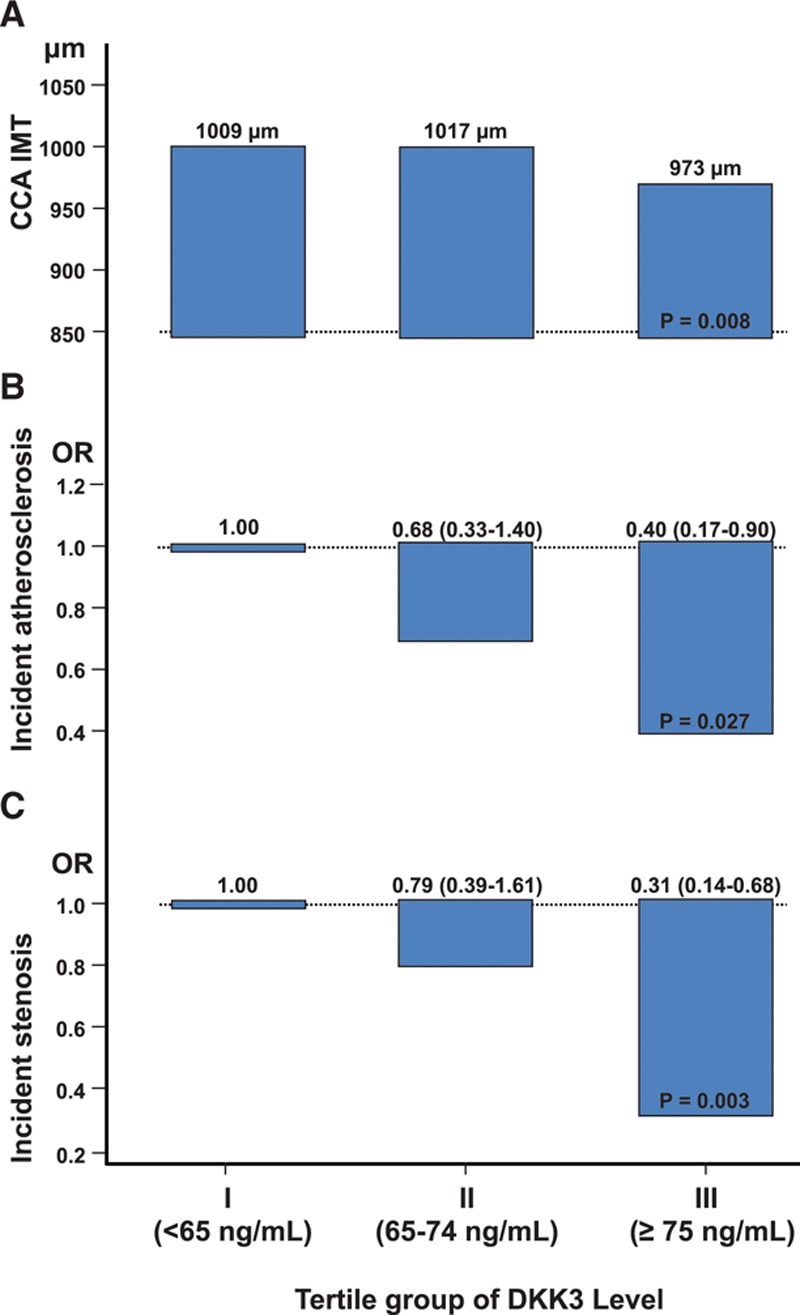
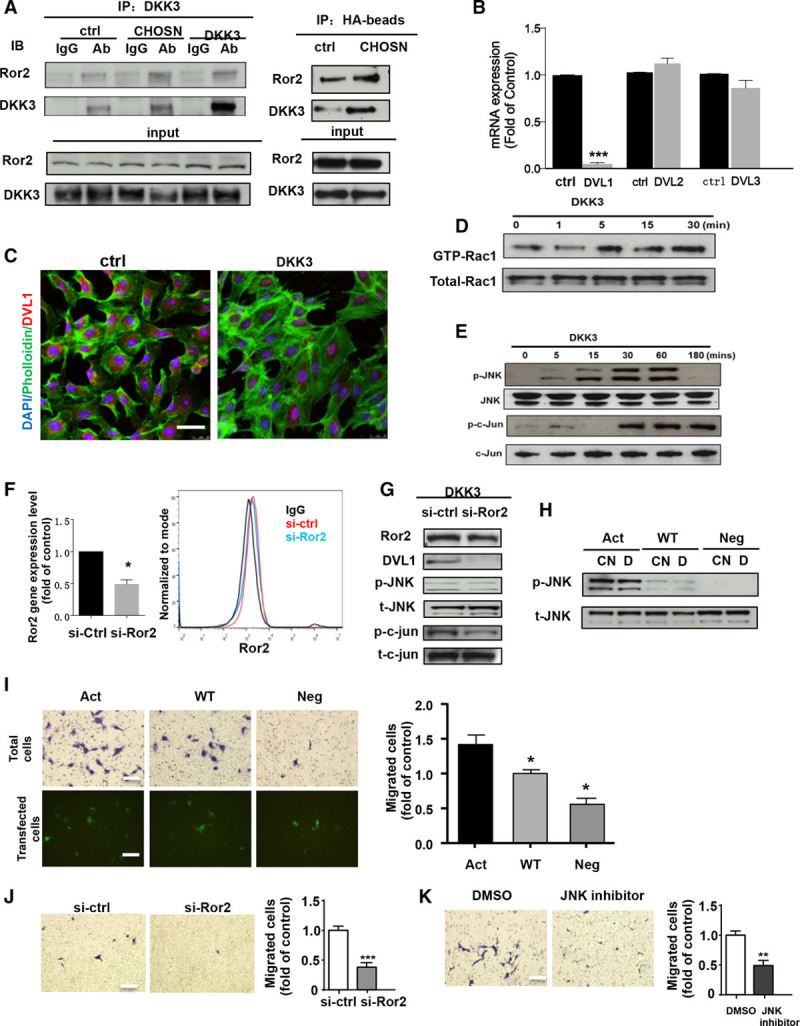
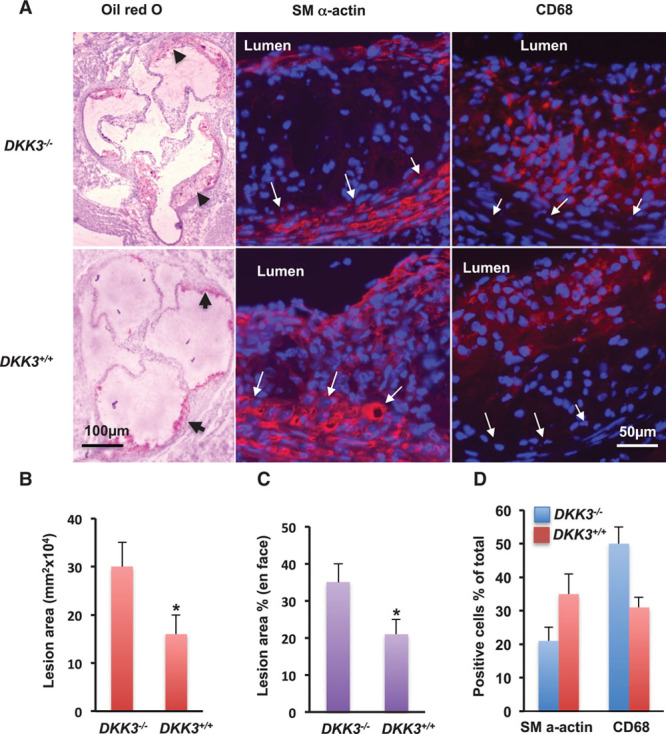
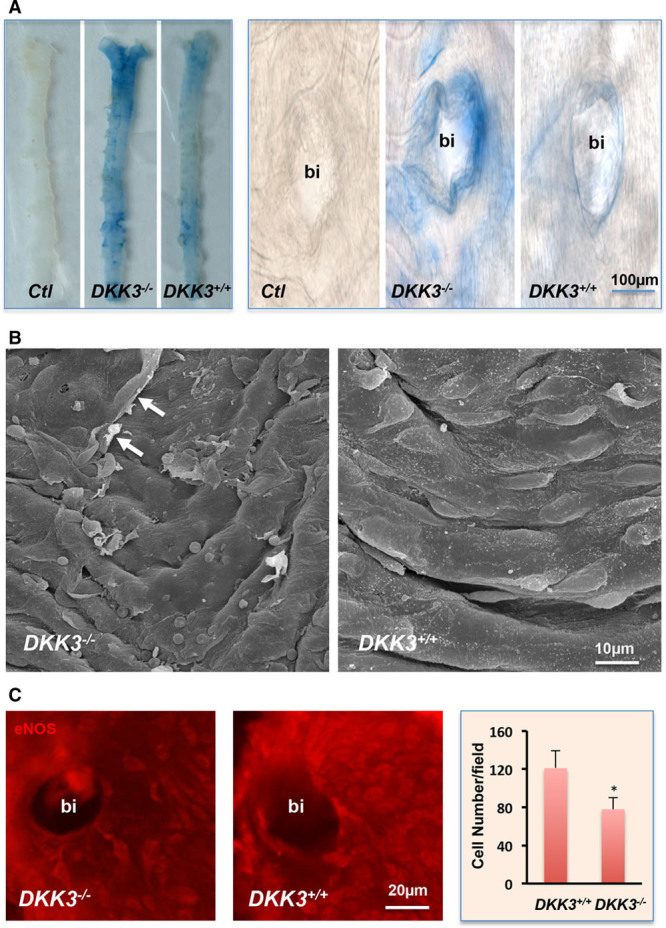
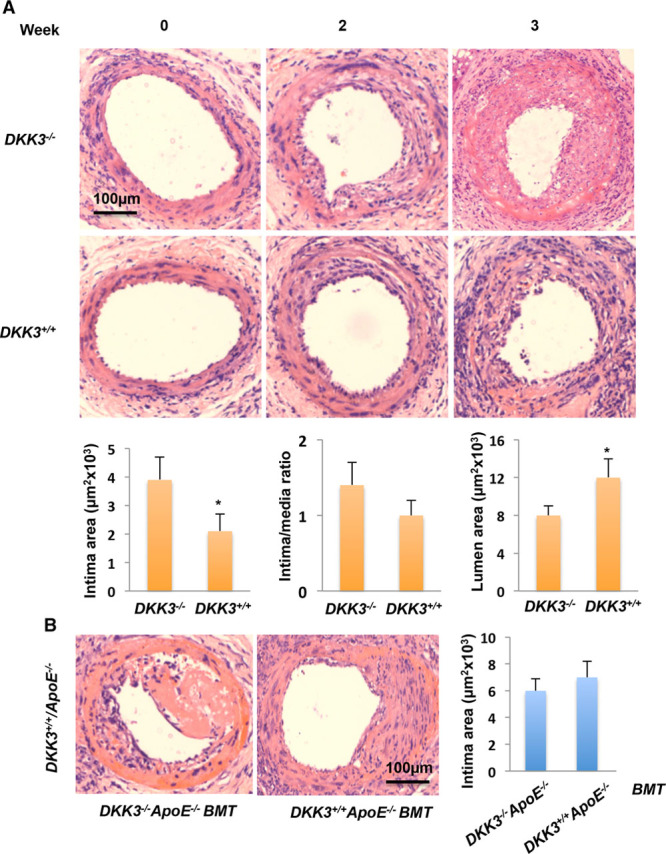
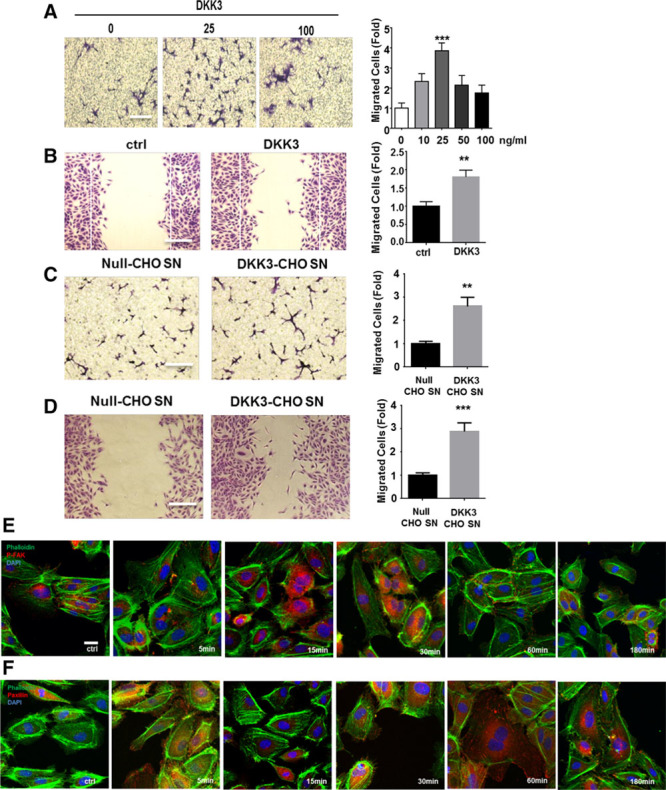
In the prospective population-based Bruneck Study, the level of plasma DKK3 was inversely related to carotid artery intima-media thickness and 5-year progression of carotid atherosclerosis independently from standard risk factors for atherosclerosis. Experimentally, we analyzed the area of atherosclerotic lesions, femoral artery injury-induced reendothelialization, and neointima formation in both and mice. It was demonstrated that DKK3 deficiency accelerated atherosclerosis and delayed reendothelialization with consequently exacerbated neointima formation. To explore the underlying mechanisms, we performed transwell and scratch migration assays using cultured human endothelial cells, which exhibited a significant induction in cell migration in response to DKK3 stimulation. This DKK3-induced migration activated ROR2 and DVL1, activated Rac1 GTPases, and upregulated JNK and c-jun phosphorylation in endothelial cells. Knockdown of the ROR2 receptor using specific siRNA or transfection of a dominant-negative form of Rac1 in endothelial cells markedly inhibited cell migration and downstream JNK and c-jun phosphorylation.
This study provides the evidence for a role of DKK3 in the protection against atherosclerosis involving endothelial migration and repair, with great therapeutic potential implications against atherosclerosis.
Dickkopf相关蛋白3(DKK3)是一种分泌蛋白,参与心脏重塑和血管平滑肌细胞分化的调节,但对其在动脉粥样硬化中的作用知之甚少。
我们使用流行病学和实验方法检验了DKK3具有抗动脉粥样硬化作用的假设。在2000年的布伦内克研究中测量了684名受试者的血液DKK3水平,随后在2005年对574名受试者进行了测量。将DKK3基因缺陷小鼠与其他小鼠杂交,以评估动脉粥样硬化的发展和血管损伤诱导的内膜形成。使用体外细胞培养模型研究内皮细胞迁移及其潜在机制。
在基于人群的前瞻性布伦内克研究中,血浆DKK3水平与颈动脉内膜中层厚度以及颈动脉粥样硬化的5年进展呈负相关,且独立于动脉粥样硬化的标准危险因素。在实验中,我们分析了DKK3基因缺陷小鼠和正常小鼠的动脉粥样硬化病变面积、股动脉损伤诱导的再内皮化和内膜形成。结果表明,DKK3基因缺陷会加速动脉粥样硬化并延迟再内皮化,从而加剧内膜形成。为了探究潜在机制,我们使用培养的人内皮细胞进行了Transwell和划痕迁移试验,结果显示DKK3刺激可显著诱导细胞迁移。这种由DKK3诱导的迁移激活了ROR2和DVL1,激活了Rac1 GTP酶,并上调了内皮细胞中JNK和c-jun的磷酸化。使用特异性siRNA敲低ROR2受体或在内皮细胞中转染显性负性形式的Rac1可显著抑制细胞迁移以及下游JNK和c-jun的磷酸化。
本研究为DKK3在预防动脉粥样硬化中发挥作用提供了证据,其涉及内皮迁移和修复,对动脉粥样硬化具有巨大的治疗潜在意义。