Vlaisavljevich Bess, Odoh Samuel O, Schnell Sondre K, Dzubak Allison L, Lee Kyuho, Planas Nora, Neaton Jeffrey B, Gagliardi Laura, Smit Berend
Department of Chemical and Biomolecular Engineering , University of California , 201 Gilman Hall , Berkeley , California 94720 , USA . Email:
Department of Chemistry , Chemical Theory Center and Supercomputing Institute , University of Minnesota , Minneapolis , Minnesota 55455-0431 , USA . Email:
Chem Sci. 2015 Sep 1;6(9):5177-5185. doi: 10.1039/c5sc01828e. Epub 2015 Jun 17.
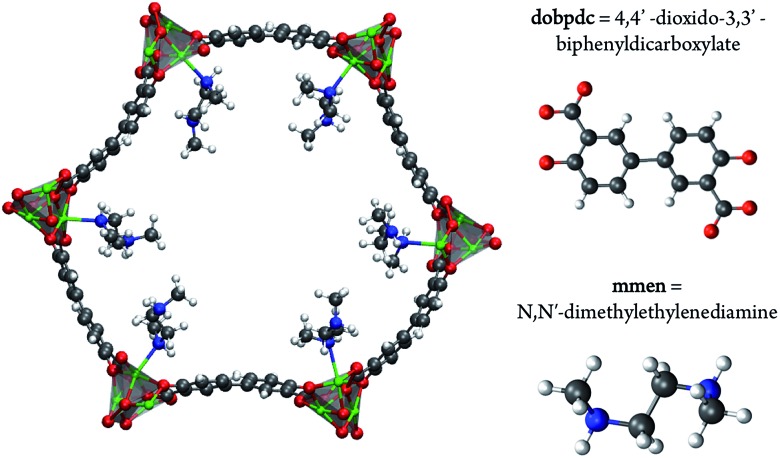
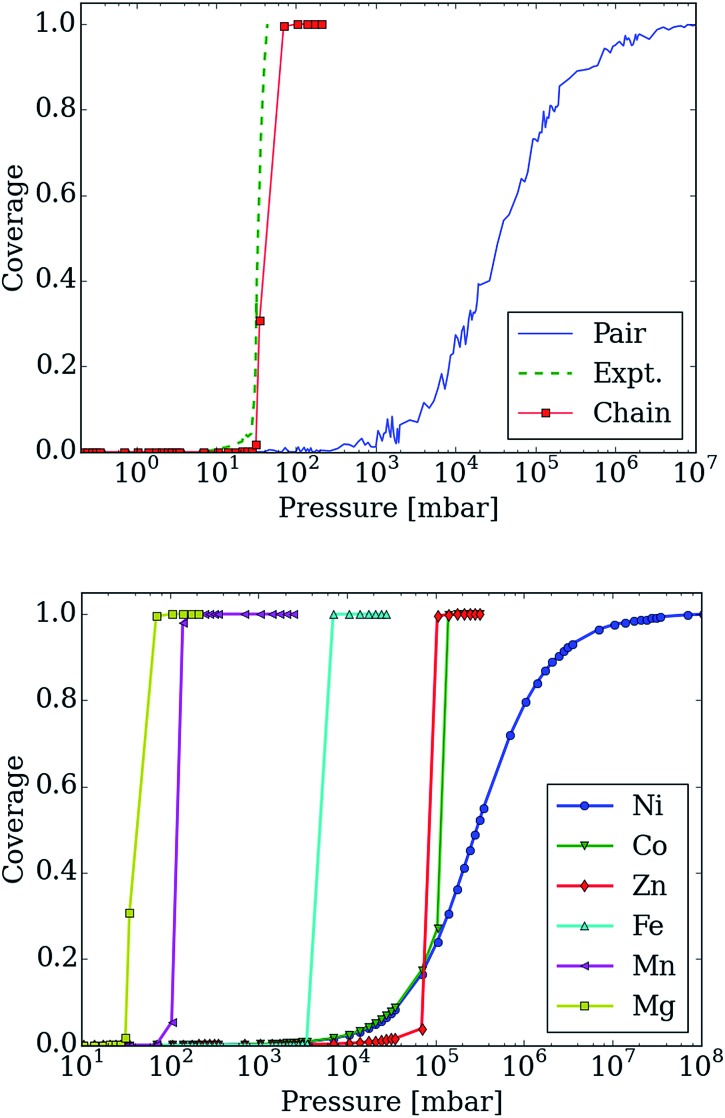
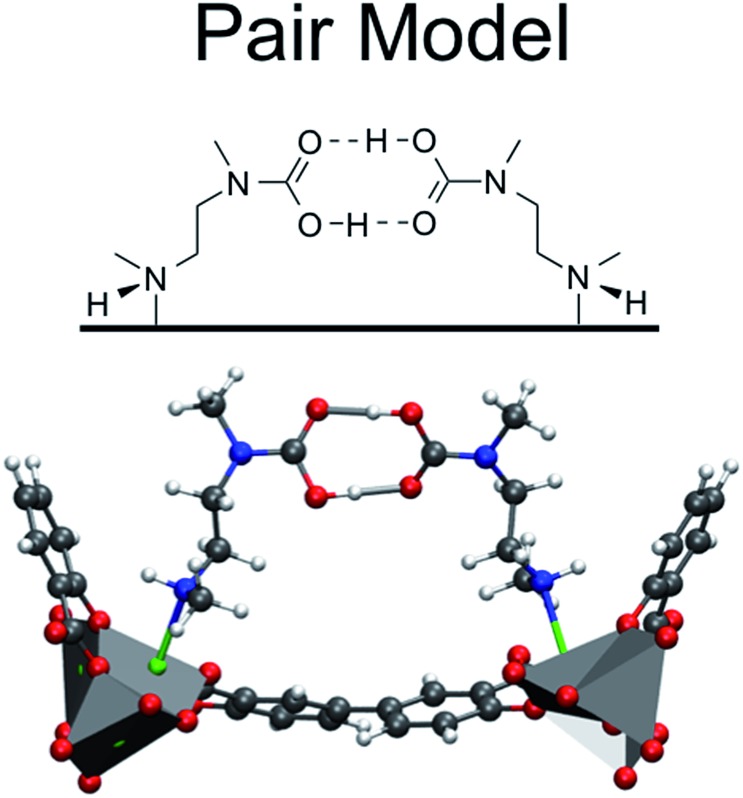
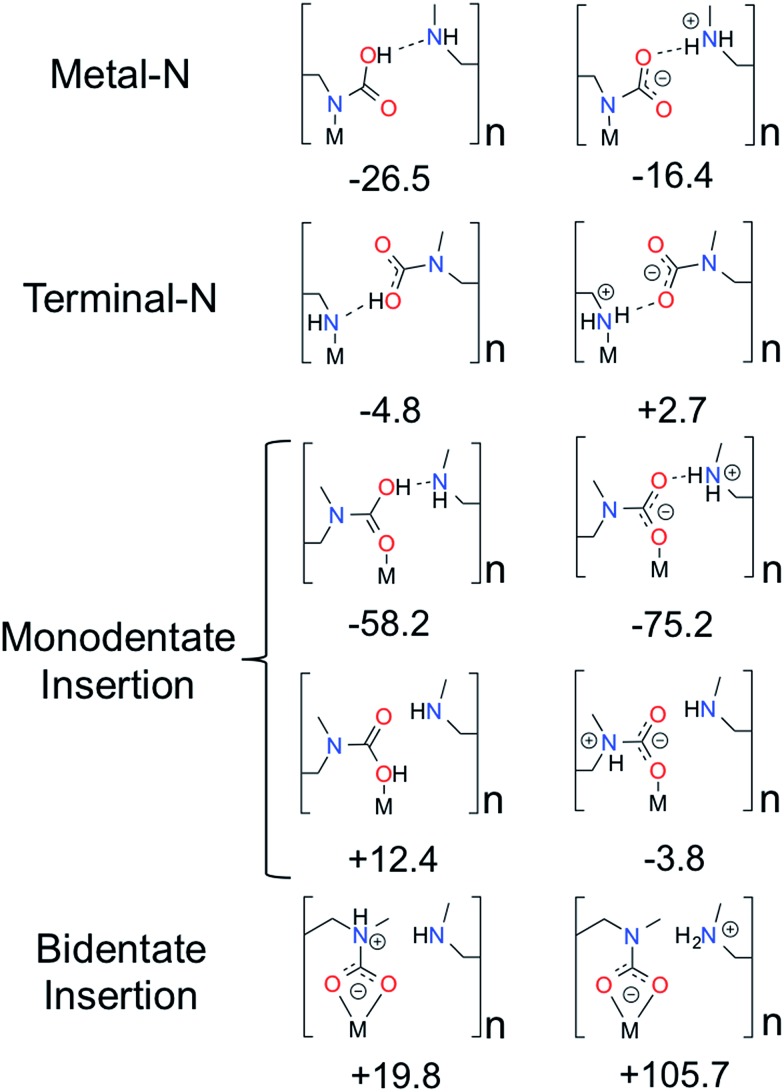
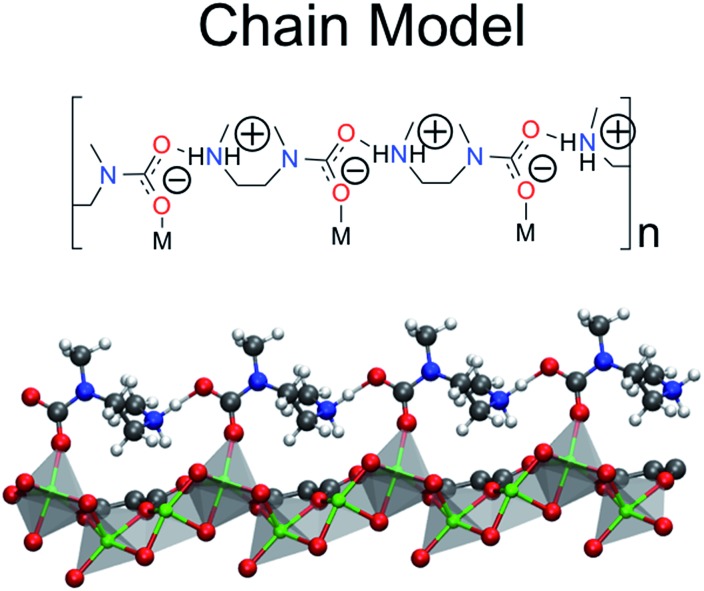
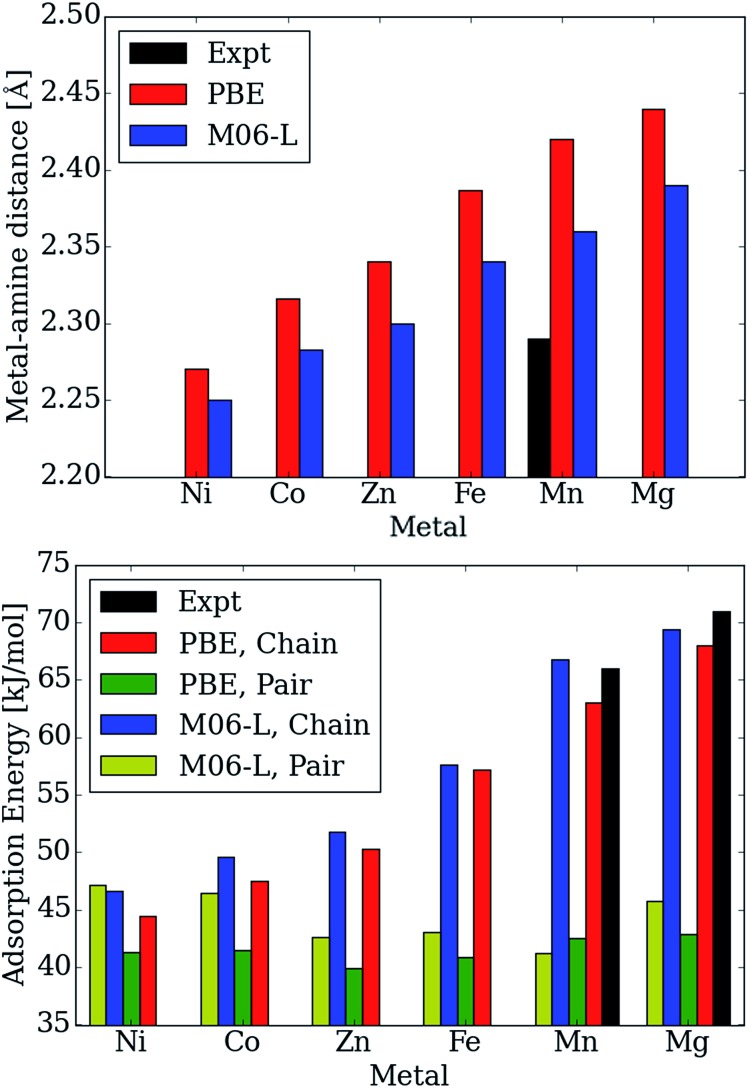
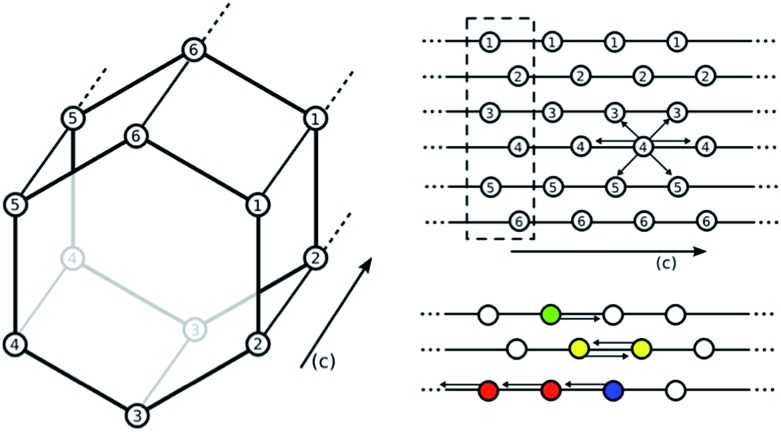
Using a combination of density functional theory and lattice models, we study the effect of CO adsorption in an amine functionalized metal-organic framework. These materials exhibit a step in the adsorption isotherm indicative of a phase change. The pressure at which this step occurs is not only temperature dependent but is also metal center dependent. Likewise, the heats of adsorption vary depending on the metal center. Herein we demonstrate quantum chemical calculations that the amines should not be considered firmly anchored to the framework and we explore the mechanism for CO adsorption. An ammonium carbamate species is formed the insertion of CO into the M-N bonds. Furthermore, we translate the quantum chemical results into isotherms using a coarse grained Monte Carlo simulation technique and show that this adsorption mechanism can explain the characteristic step observed in the experimental isotherm while a previously proposed mechanism cannot. Furthermore, metal analogues have been explored and the CO binding energies show a strong metal dependence corresponding to the M-N bond strength. We show that this difference can be exploited to tune the pressure at which the step in the isotherm occurs. Additionally, the mmen-Ni(dobpdc) framework shows Langmuir like behavior, and our simulations show how this can be explained by competitive adsorption between the new model and a previously proposed model.
我们使用密度泛函理论和晶格模型相结合的方法,研究了胺功能化金属有机框架中CO吸附的影响。这些材料在吸附等温线上呈现出一个表明相变的台阶。这个台阶出现的压力不仅取决于温度,还取决于金属中心。同样,吸附热也因金属中心而异。在此,我们通过量子化学计算证明胺不应被视为牢固地锚定在框架上,并探索了CO吸附的机制。通过CO插入M-N键形成了氨基甲酸铵物种。此外,我们使用粗粒化蒙特卡罗模拟技术将量子化学结果转化为等温线,并表明这种吸附机制可以解释实验等温线中观察到的特征台阶,而先前提出的机制则无法解释。此外,还探索了金属类似物,CO结合能显示出与M-N键强度相对应的强烈金属依赖性。我们表明,可以利用这种差异来调节等温线中台阶出现的压力。此外,mmen-Ni(dobpdc)框架表现出类似朗缪尔的行为,我们的模拟展示了如何通过新模型与先前提出的模型之间的竞争吸附来解释这一现象。