UK Dementia Research Institute, University of Edinburgh, Edinburgh, United Kingdom.
Edinburgh Medical School, University of Edinburgh, Edinburgh, United Kingdom.
Elife. 2017 Jul 21;6:e17161. doi: 10.7554/eLife.17161.
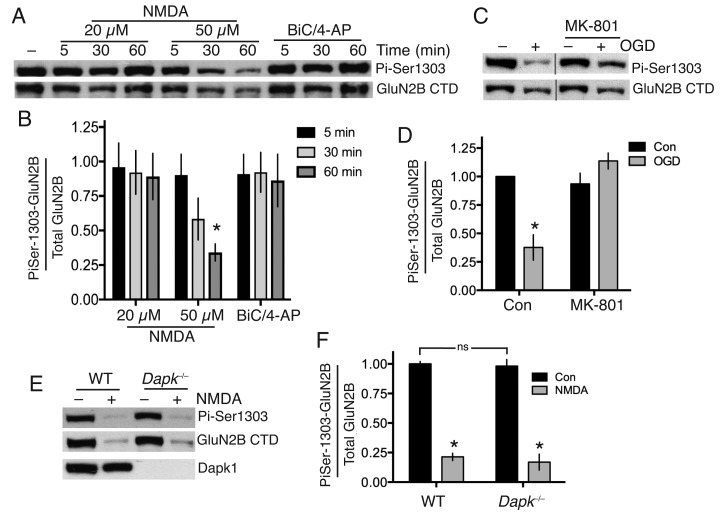
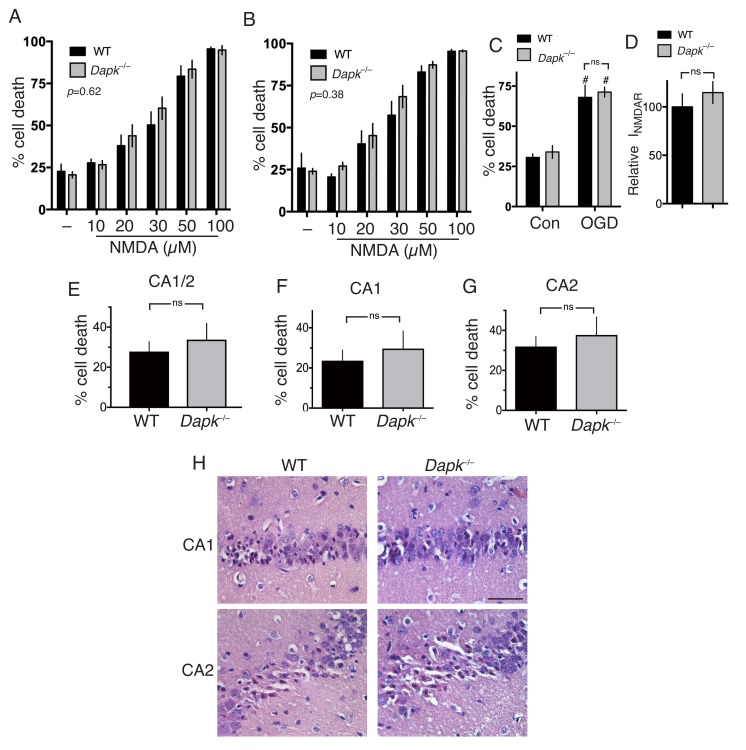
Aberrant NMDA receptor (NMDAR) activity contributes to several neurological disorders, but direct antagonism is poorly tolerated therapeutically. The GluN2B cytoplasmic C-terminal domain (CTD) represents an alternative therapeutic target since it potentiates excitotoxic signaling. The key GluN2B CTD-centred event in excitotoxicity is proposed to involve its phosphorylation at Ser-1303 by Dapk1, that is blocked by a neuroprotective cell-permeable peptide mimetic of the region. Contrary to this model, we find that excitotoxicity can proceed without increased Ser-1303 phosphorylation, and is unaffected by Dapk1 deficiency in vitro or following ischemia in vivo. Pharmacological analysis of the aforementioned neuroprotective peptide revealed that it acts in a sequence-independent manner as an open-channel NMDAR antagonist at or near the Mg site, due to its high net positive charge. Thus, GluN2B-driven excitotoxic signaling can proceed independently of Dapk1 or altered Ser-1303 phosphorylation.
异常的 NMDA 受体(NMDAR)活性与几种神经疾病有关,但直接拮抗在治疗上的耐受性较差。GluN2B 细胞质 C 末端结构域(CTD)代表了一种替代的治疗靶点,因为它增强了兴奋性毒性信号。兴奋性毒性中的关键 GluN2B CTD 中心事件据推测涉及 Dapk1 对其 Ser-1303 的磷酸化,而该磷酸化被该区域的神经保护细胞可渗透肽模拟物所阻断。与该模型相反,我们发现兴奋性毒性可以在没有增加 Ser-1303 磷酸化的情况下进行,并且在体外 Dapk1 缺乏或体内缺血后不受影响。对上述神经保护肽的药理学分析表明,由于其净正电荷高,它以序列非依赖性方式作为位于或靠近 Mg 位点的开放通道 NMDAR 拮抗剂发挥作用。因此,GluN2B 驱动的兴奋性毒性信号可以独立于 Dapk1 或改变的 Ser-1303 磷酸化进行。