Park Soomin, Baek Seung-Hun, Cho Sang-Nae, Jang Young-Saeng, Kim Ahreum, Choi In-Hong
Department of Microbiology, Institute for Immunology and Immunological Diseases, and Brain Korea 21 PLUS Project for Medical Science, Yonsei University College of MedicineSeoul, South Korea.
Front Cell Infect Microbiol. 2017 Jul 13;7:314. doi: 10.3389/fcimb.2017.00314. eCollection 2017.
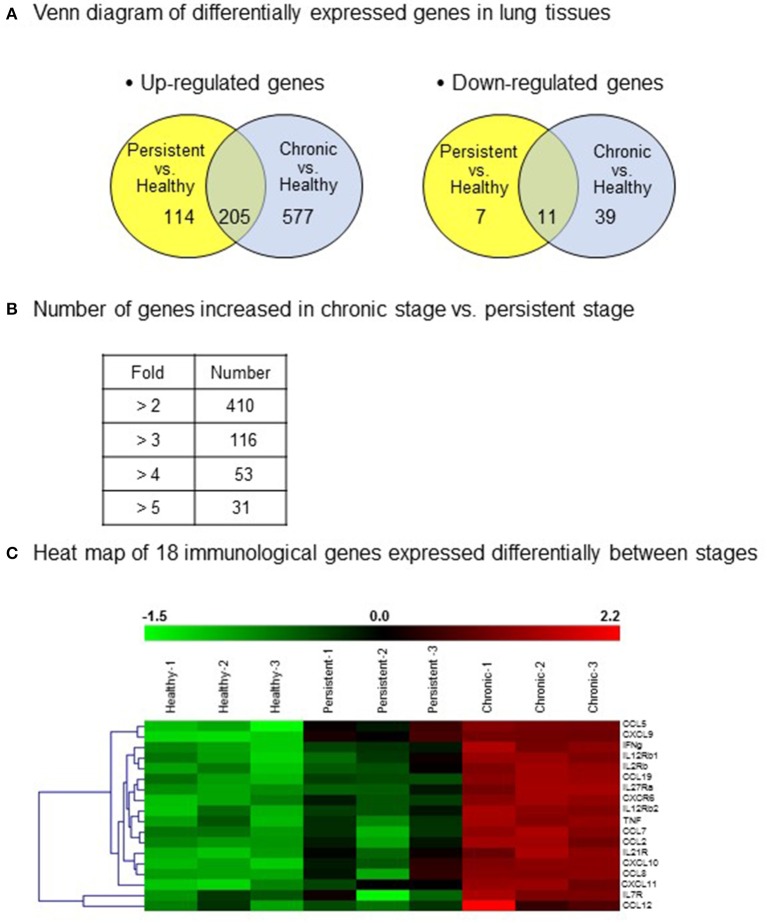
There is a substantial need for biomarkers to distinguish latent stage from active infections, for predicting disease progression. To induce the reactivation of tuberculosis, we present a new experimental animal model modified based on the previous model established by our group. In the new model, the reactivation of tuberculosis is induced without administration of immunosuppressive agents, which might disturb immune responses. To identify the immunological status of the persistent and chronic stages, we analyzed immunological genes in lung tissues from mice infected with . Gene expression was screened using cDNA microarray analysis and confirmed by quantitative RT-PCR. Based on the cDNA microarray results, 11 candidate cytokines genes, which were obviously up-regulated during the chronic stage compared with those during the persistent stage, were selected and clustered into three groups: (1) chemokine genes, except those of monocyte chemoattractant proteins (MCPs; CXCL9, CXCL10, CXCL11, CCL5, CCL19); (2) MCP genes (CCL2, CCL7, CCL8, CCL12); and (3) TNF and IFN-γ genes. Results from the cDNA microarray and quantitative RT-PCR analyses revealed that the mRNA expression of the selected cytokine genes was significantly higher in lung tissues of the chronic stage than of the persistent stage. Three chemokines (CCL5, CCL19, and CXCL9) and three MCPs (CCL7, CCL2, and CCL12) were noticeably increased in the chronic stage compared with the persistent stage by cDNA microarray ( < 0.01, except CCL12) or RT-PCR ( < 0.01). Therefore, these six significantly increased cytokines in lung tissue from the mouse tuberculosis model might be candidates for biomarkers to distinguish the two disease stages. This information can be combined with already reported potential biomarkers to construct a network of more efficient tuberculosis markers.
迫切需要生物标志物来区分潜伏阶段和活动性感染,以预测疾病进展。为了诱导结核病的再激活,我们提出了一种基于本课题组之前建立的模型改进的新实验动物模型。在新模型中,无需施用可能干扰免疫反应的免疫抑制剂即可诱导结核病的再激活。为了确定持续性和慢性阶段的免疫状态,我们分析了感染小鼠肺组织中的免疫基因。使用cDNA微阵列分析筛选基因表达,并通过定量RT-PCR进行确认。基于cDNA微阵列结果,选择了11个候选细胞因子基因,这些基因在慢性阶段与持续性阶段相比明显上调,并聚类为三组:(1)趋化因子基因,但不包括单核细胞趋化蛋白(MCPs;CXCL9、CXCL10、CXCL11、CCL5、CCL19);(2)MCP基因(CCL2、CCL7、CCL8、CCL12);(3)TNF和IFN-γ基因。cDNA微阵列和定量RT-PCR分析结果显示,所选细胞因子基因的mRNA表达在慢性阶段的肺组织中显著高于持续性阶段。与持续性阶段相比,通过cDNA微阵列(除CCL12外,P<0.01)或RT-PCR(P<0.01)检测,慢性阶段有三种趋化因子(CCL5、CCL19和CXCL9)和三种MCPs(CCL7、CCL2和CCL12)明显增加。因此,小鼠结核病模型肺组织中这六种显著增加的细胞因子可能是区分两个疾病阶段的生物标志物候选物。这些信息可以与已报道的潜在生物标志物相结合,构建一个更有效的结核病标志物网络。