Mademont-Soler Irene, Mates Jesus, Yotti Raquel, Espinosa Maria Angeles, Pérez-Serra Alexandra, Fernandez-Avila Ana Isabel, Coll Monica, Méndez Irene, Iglesias Anna, Del Olmo Bernat, Riuró Helena, Cuenca Sofía, Allegue Catarina, Campuzano Oscar, Picó Ferran, Ferrer-Costa Carles, Álvarez Patricia, Castillo Sergio, Garcia-Pavia Pablo, Gonzalez-Lopez Esther, Padron-Barthe Laura, Díaz de Bustamante Aranzazu, Darnaude María Teresa, González-Hevia José Ignacio, Brugada Josep, Fernandez-Aviles Francisco, Brugada Ramon
Cardiovascular Genetics Center, University of Girona-IDIBGI, Girona, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), Madrid, Spain.
PLoS One. 2017 Aug 3;12(8):e0181465. doi: 10.1371/journal.pone.0181465. eCollection 2017.
Hypertrophic cardiomyopathy (HCM) is the most prevalent inherited heart disease. Next-generation sequencing (NGS) is the preferred genetic test, but the diagnostic value of screening for minor and candidate genes, and the role of copy number variants (CNVs) deserves further evaluation.
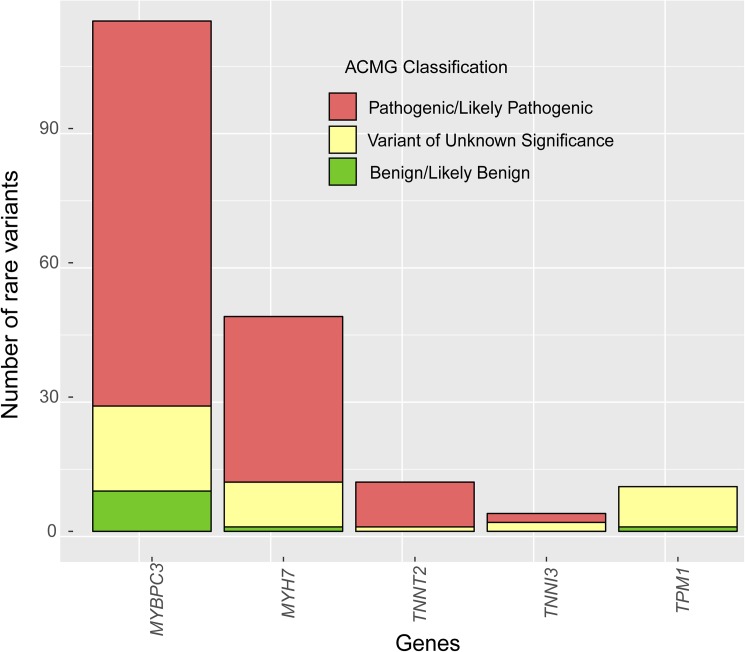
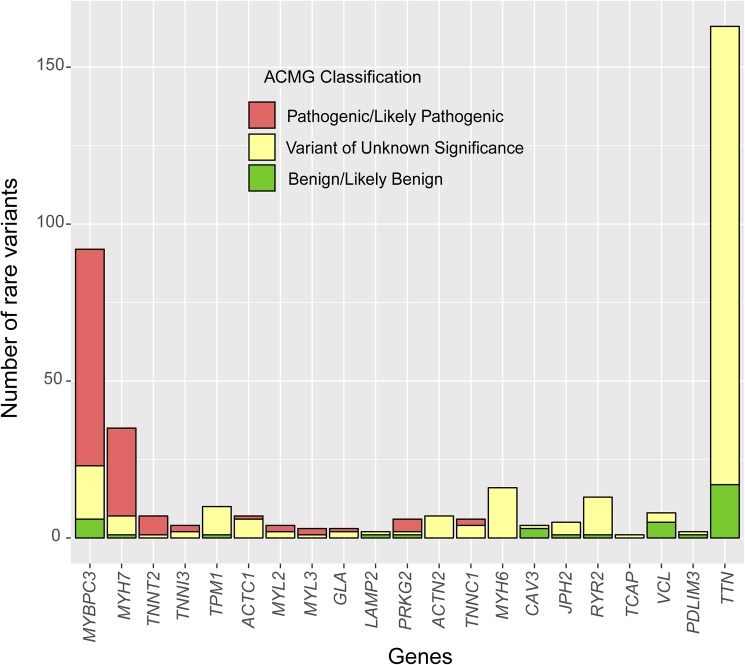
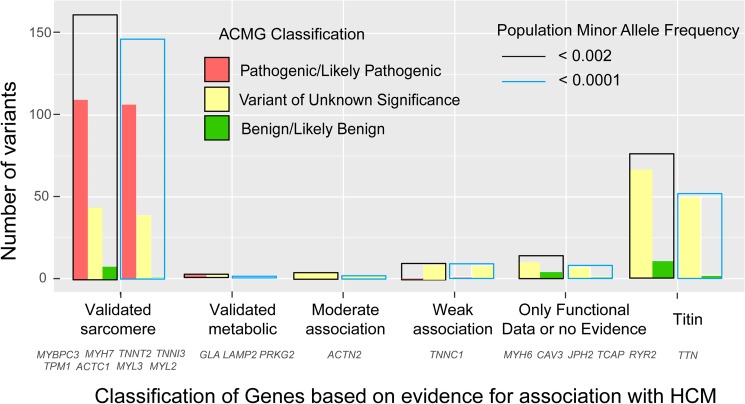
Three hundred and eighty-seven consecutive unrelated patients with HCM were screened for genetic variants in the 5 most frequent genes (MYBPC3, MYH7, TNNT2, TNNI3 and TPM1) using Sanger sequencing (N = 84) or NGS (N = 303). In the NGS cohort we analyzed 20 additional minor or candidate genes, and applied a proprietary bioinformatics algorithm for detecting CNVs. Additionally, the rate and classification of TTN variants in HCM were compared with 427 patients without structural heart disease.
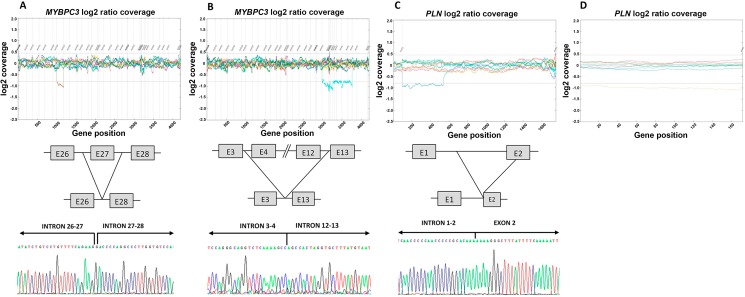
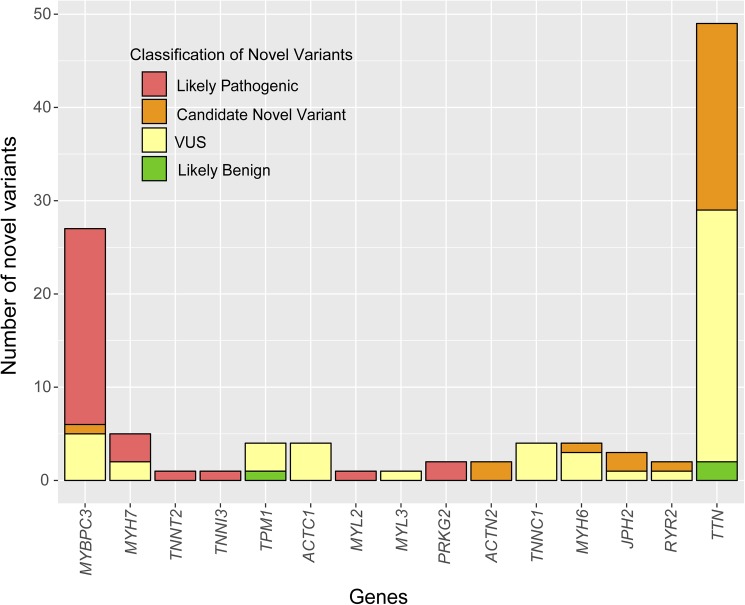
The percentage of patients with pathogenic/likely pathogenic (P/LP) variants in the main genes was 33.3%, without significant differences between the Sanger sequencing and NGS cohorts. The screening for 20 additional genes revealed LP variants in ACTC1, MYL2, MYL3, TNNC1, GLA and PRKAG2 in 12 patients. This approach resulted in more inconclusive tests (36.0% vs. 9.6%, p<0.001), mostly due to variants of unknown significance (VUS) in TTN. The detection rate of rare variants in TTN was not significantly different to that found in the group of patients without structural heart disease. In the NGS cohort, 4 patients (1.3%) had pathogenic CNVs: 2 deletions in MYBPC3 and 2 deletions involving the complete coding region of PLN.
A small percentage of HCM cases without point mutations in the 5 main genes are explained by P/LP variants in minor or candidate genes and CNVs. Screening for variants in TTN in HCM patients drastically increases the number of inconclusive tests, and shows a rate of VUS that is similar to patients without structural heart disease, suggesting that this gene should not be analyzed for clinical purposes in HCM.
肥厚型心肌病(HCM)是最常见的遗传性心脏病。二代测序(NGS)是首选的基因检测方法,但筛查次要基因和候选基因的诊断价值以及拷贝数变异(CNV)的作用值得进一步评估。
对387例连续的无亲缘关系的HCM患者,使用桑格测序法(N = 84)或NGS(N = 303)筛查5个最常见基因(MYBPC3、MYH7、TNNT2、TNNI3和TPM1)中的基因变异。在NGS队列中,我们分析了另外20个次要或候选基因,并应用一种专有的生物信息学算法检测CNV。此外,将HCM患者中TTN变异的发生率和分类与427例无结构性心脏病的患者进行比较。
主要基因中存在致病/可能致病(P/LP)变异的患者百分比为33.3%,桑格测序队列和NGS队列之间无显著差异。对另外20个基因的筛查在12例患者的ACTC1、MYL2、MYL3、TNNC1、GLA和PRKAG2中发现了LP变异。这种方法导致更多不确定的检测结果(36.0%对9.6%,p<0.001),主要是由于TTN中意义未明的变异(VUS)。TTN中罕见变异的检出率与无结构性心脏病患者组无显著差异。在NGS队列中,4例患者(1.3%)有致病CNV:2例MYBPC3缺失和2例涉及PLN完整编码区的缺失。
5个主要基因无点突变 的一小部分HCM病例可由次要或候选基因中的P/LP变异和CNV解释。对HCM患者的TTN变异进行筛查会大幅增加不确定检测的数量,并且显示出与无结构性心脏病患者相似的VUS发生率,这表明在HCM的临床诊断中不应分析该基因。