Division of Cardiocirculatory Signaling, National Institute for Physiological Sciences (Okazaki Institute for Integrative Bioscience), National Institutes of Natural Sciences, Aichi, 444-8787, Japan.
Department of Physiological Sciences, SOKENDAI (School of Life Science, The Graduate University for Advanced Studies), Aichi, 444-8787, Japan.
Sci Rep. 2017 Aug 8;7(1):7511. doi: 10.1038/s41598-017-07903-4.
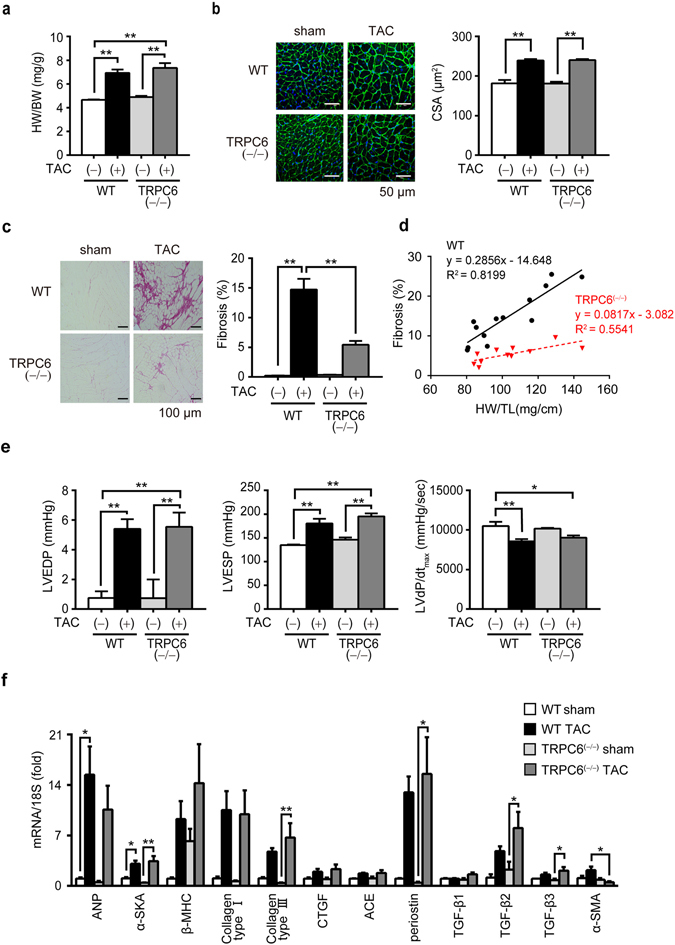
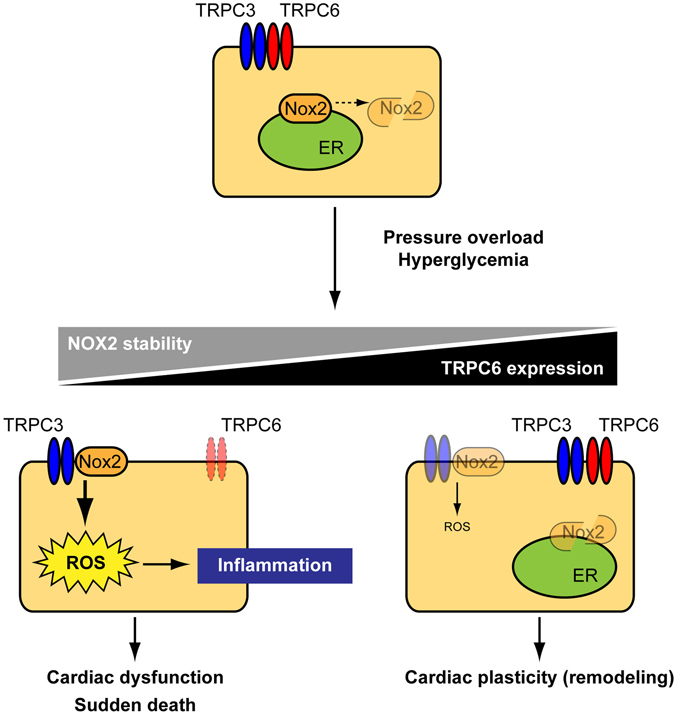
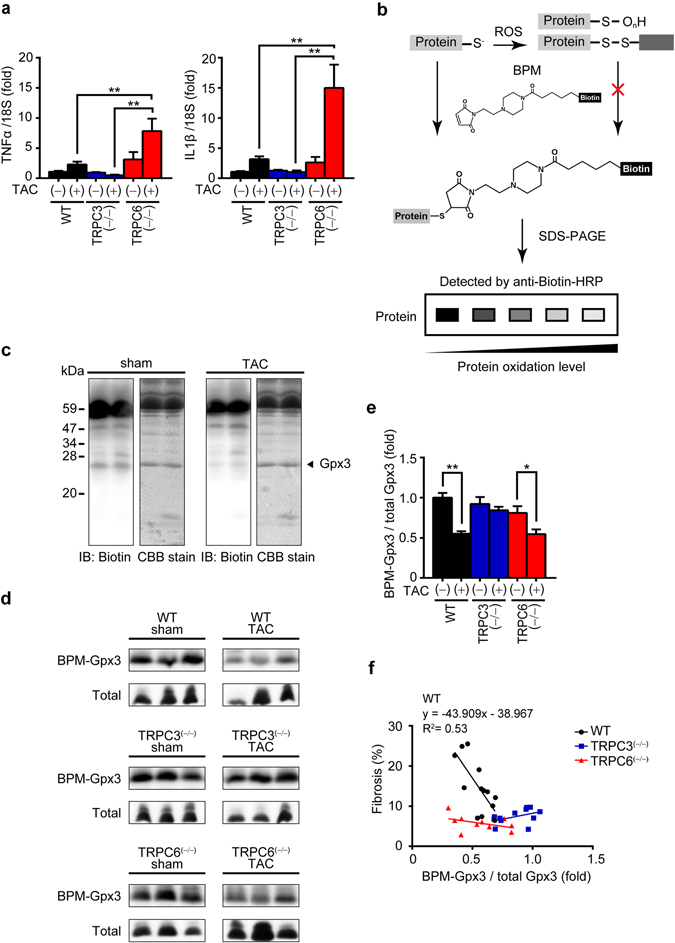
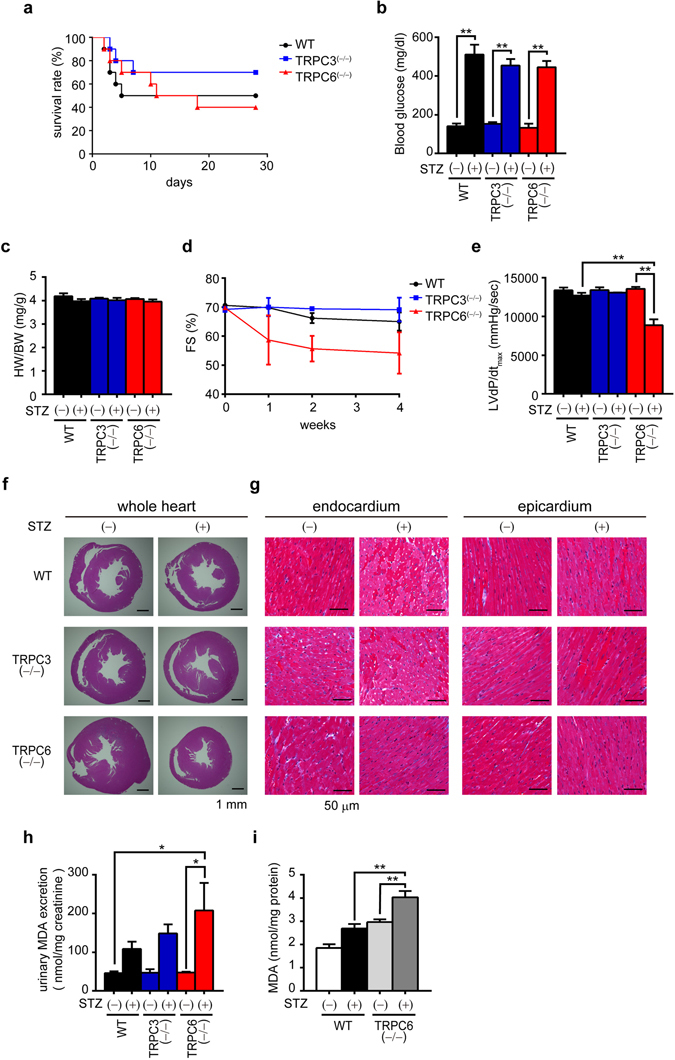
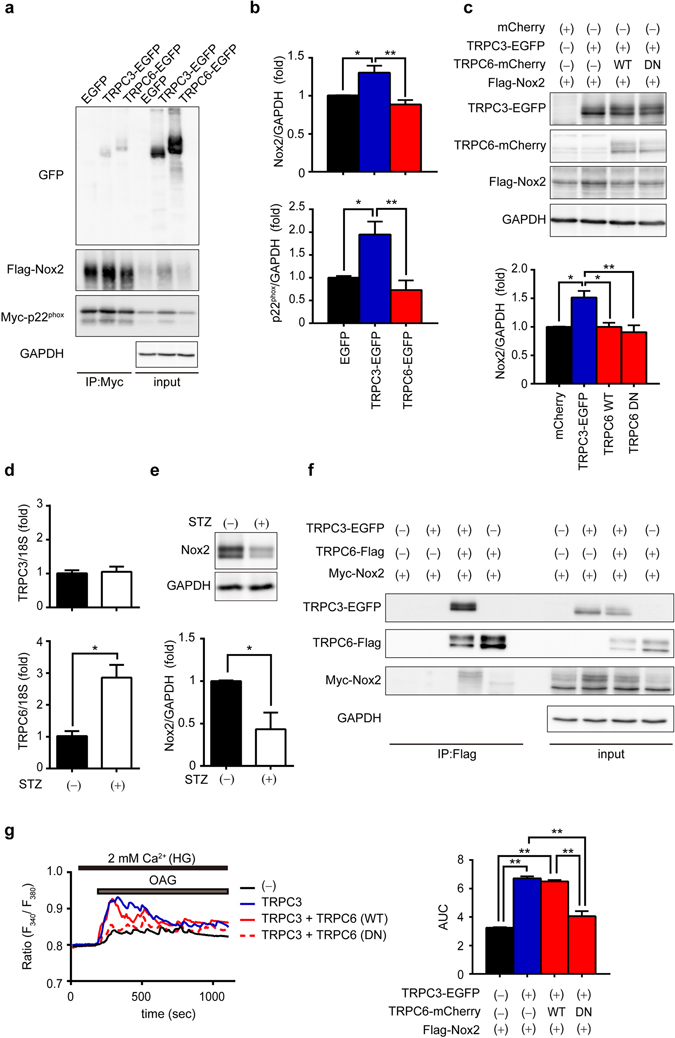
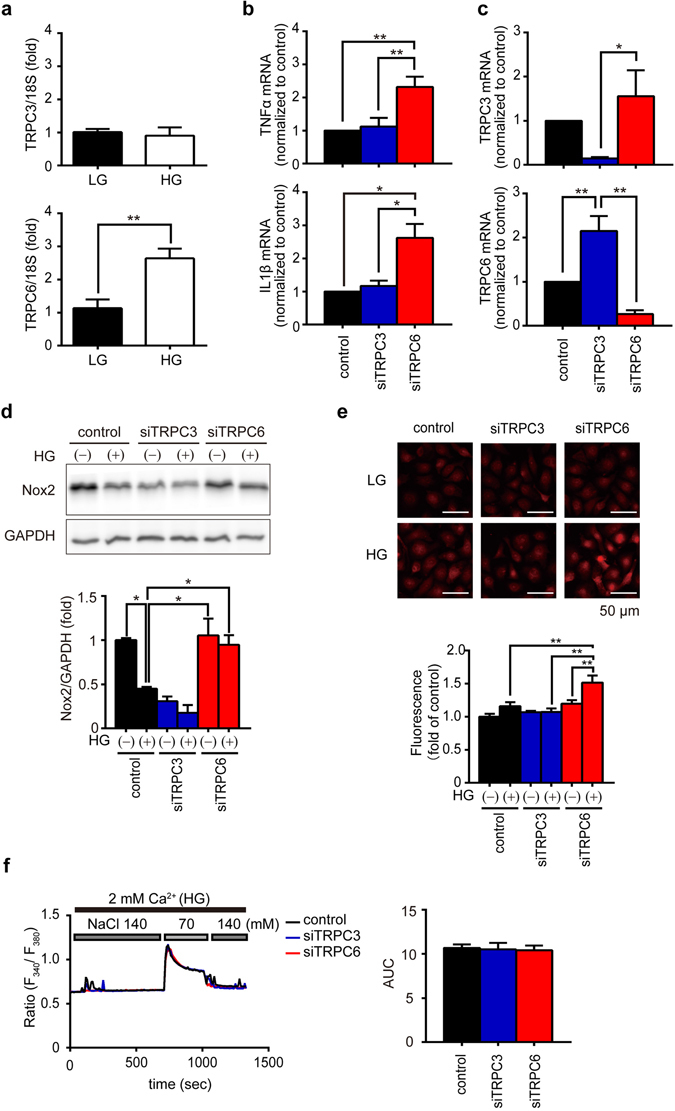
Excess production of reactive oxygen species (ROS) caused by hyperglycemia is a major risk factor for heart failure. We previously reported that transient receptor potential canonical 3 (TRPC3) channel mediates pressure overload-induced maladaptive cardiac fibrosis by forming stably functional complex with NADPH oxidase 2 (Nox2). Although TRPC3 has been long suggested to form hetero-multimer channels with TRPC6 and function as diacylglycerol-activated cation channels coordinately, the role of TRPC6 in heart is still obscure. We here demonstrated that deletion of TRPC6 had no impact on pressure overload-induced heart failure despite inhibiting interstitial fibrosis in mice. TRPC6-deficient mouse hearts 1 week after transverse aortic constriction showed comparable increases in fibrotic gene expressions and ROS production but promoted inductions of inflammatory cytokines, compared to wild type hearts. Treatment of TRPC6-deficient mice with streptozotocin caused severe reduction of cardiac contractility with enhancing urinary and cardiac lipid peroxide levels, compared to wild type and TRPC3-deficient mice. Knockdown of TRPC6, but not TRPC3, enhanced basal expression levels of cytokines in rat cardiomyocytes. TRPC6 could interact with Nox2, but the abundance of TRPC6 was inversely correlated with that of Nox2. These results strongly suggest that Nox2 destabilization through disrupting TRPC3-Nox2 complex underlies attenuation of hyperglycemia-induced heart failure by TRPC6.
高血糖引起的活性氧(ROS)过度产生是心力衰竭的一个主要危险因素。我们之前报道过,瞬时受体电位经典型 3(TRPC3)通道通过与 NADPH 氧化酶 2(Nox2)形成稳定的功能复合物,介导压力超负荷引起的适应性心脏纤维化。尽管 TRPC3 长期以来被认为与 TRPC6 形成异源多聚体通道,并作为二酰基甘油激活的阳离子通道协同发挥作用,但 TRPC6 在心脏中的作用仍然不清楚。我们在这里证明,尽管抑制了小鼠的间质纤维化,但 TRPC6 的缺失对压力超负荷引起的心力衰竭没有影响。与野生型心脏相比,TRPC6 缺陷型小鼠心脏在横主动脉缩窄 1 周后,纤维化基因表达和 ROS 产生的增加相当,但促炎细胞因子的诱导增加。与野生型和 TRPC3 缺陷型小鼠相比,用链脲佐菌素处理 TRPC6 缺陷型小鼠导致心脏收缩力严重下降,同时尿液和心脏脂质过氧化物水平升高。与 TRPC3 不同,TRPC6 的敲低增强了大鼠心肌细胞中细胞因子的基础表达水平。TRPC6 可以与 Nox2 相互作用,但 TRPC6 的丰度与 Nox2 的丰度呈负相关。这些结果强烈表明,TRPC6 通过破坏 TRPC3-Nox2 复合物导致 Nox2 失稳,从而减轻高血糖引起的心力衰竭。