Gelfman Sahar, Wang Quanli, McSweeney K Melodi, Ren Zhong, La Carpia Francesca, Halvorsen Matt, Schoch Kelly, Ratzon Fanni, Heinzen Erin L, Boland Michael J, Petrovski Slavé, Goldstein David B
Institute for Genomic Medicine, Columbia University Medical Center, New York, New York, 10032, USA.
Department of Genetics and Development, Columbia University Medical Center, New York, New York, 10032, USA.
Nat Commun. 2017 Aug 9;8(1):236. doi: 10.1038/s41467-017-00141-2.
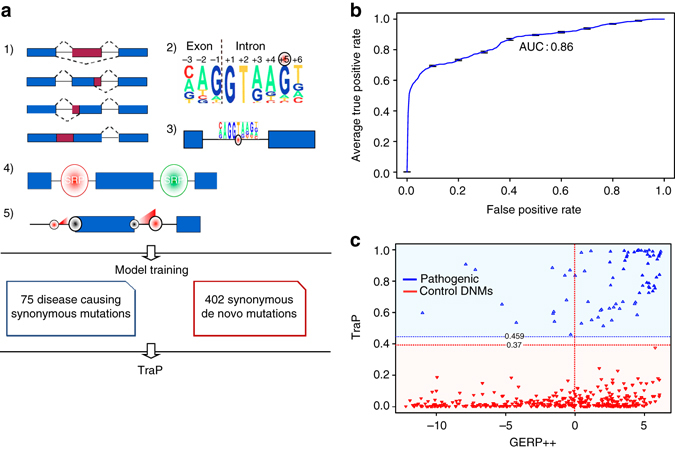
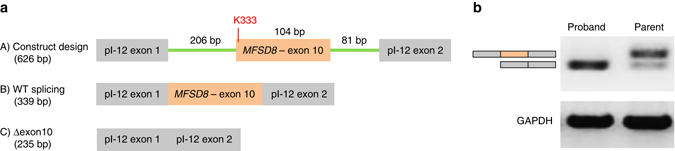
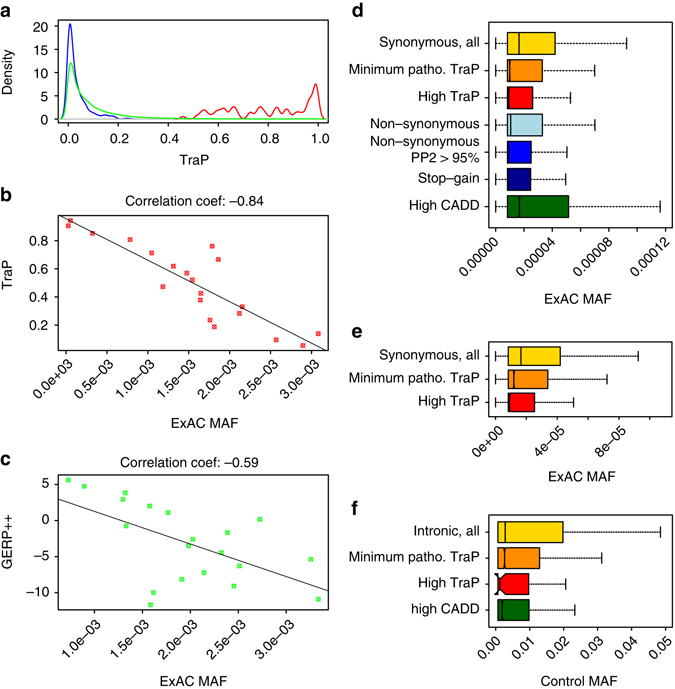
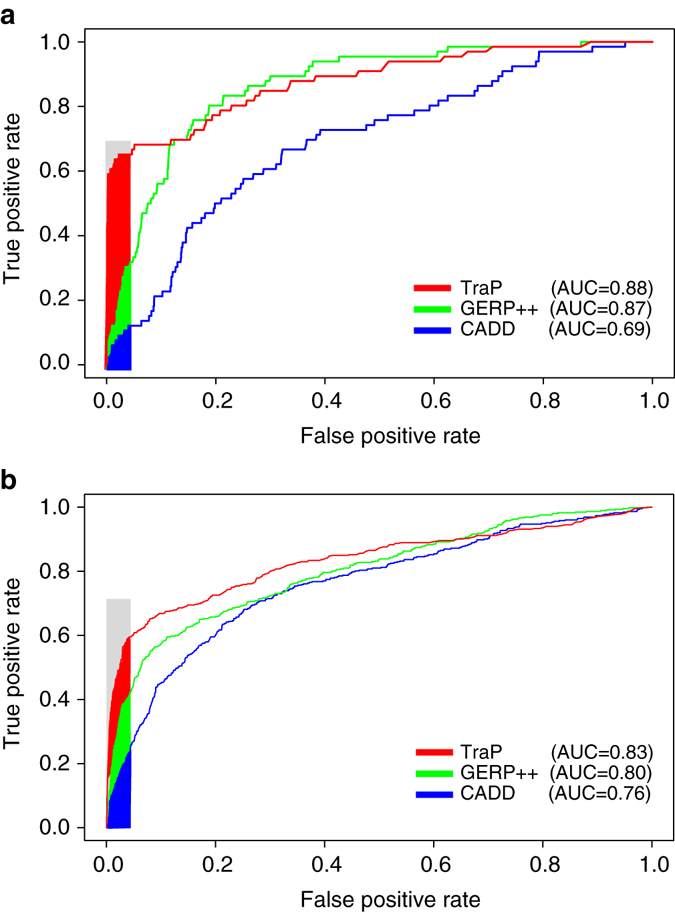
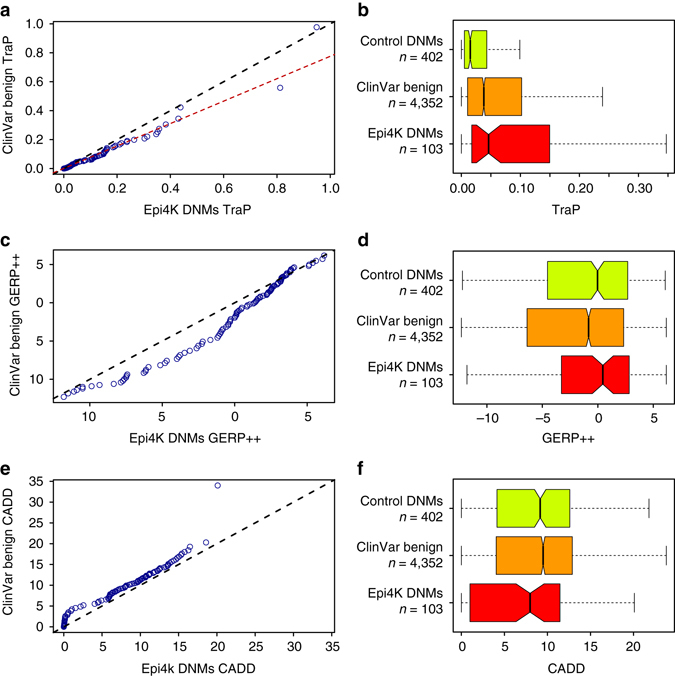
Identifying the underlying causes of disease requires accurate interpretation of genetic variants. Current methods ineffectively capture pathogenic non-coding variants in genic regions, resulting in overlooking synonymous and intronic variants when searching for disease risk. Here we present the Transcript-inferred Pathogenicity (TraP) score, which uses sequence context alterations to reliably identify non-coding variation that causes disease. High TraP scores single out extremely rare variants with lower minor allele frequencies than missense variants. TraP accurately distinguishes known pathogenic and benign variants in synonymous (AUC = 0.88) and intronic (AUC = 0.83) public datasets, dismissing benign variants with exceptionally high specificity. TraP analysis of 843 exomes from epilepsy family trios identifies synonymous variants in known epilepsy genes, thus pinpointing risk factors of disease from non-coding sequence data. TraP outperforms leading methods in identifying non-coding variants that are pathogenic and is therefore a valuable tool for use in gene discovery and the interpretation of personal genomes.While non-coding synonymous and intronic variants are often not under strong selective constraint, they can be pathogenic through affecting splicing or transcription. Here, the authors develop a score that uses sequence context alterations to predict pathogenicity of synonymous and non-coding genetic variants, and provide a web server of pre-computed scores.
识别疾病的潜在病因需要对基因变异进行准确解读。目前的方法无法有效捕捉基因区域中的致病性非编码变异,导致在寻找疾病风险时忽略了同义变异和内含子变异。在此,我们提出了转录本推断致病性(TraP)评分,它利用序列上下文改变来可靠地识别导致疾病的非编码变异。高TraP评分能筛选出次要等位基因频率比错义变异更低的极其罕见的变异。TraP在同义(AUC = 0.88)和内含子(AUC = 0.83)公共数据集中能准确区分已知的致病性变异和良性变异,以极高的特异性排除良性变异。对来自癫痫家系三联体样本的843个外显子进行TraP分析,可识别出已知癫痫基因中的同义变异,从而从非编码序列数据中确定疾病的风险因素。在识别致病性非编码变异方面,TraP优于领先的方法,因此是基因发现和个人基因组解读中的一个有价值的工具。虽然非编码同义变异和内含子变异通常不受强选择约束,但它们可通过影响剪接或转录而具有致病性。在此,作者开发了一种利用序列上下文改变来预测同义及非编码基因变异致病性的评分方法,并提供了一个预先计算好评分的网络服务器。