Nishihara Hideaki, Omoto Masatoshi, Takao Masaki, Higuchi Yujiro, Koga Michiaki, Kawai Motoharu, Kawano Hiroo, Ikeda Eiji, Takashima Hiroshi, Kanda Takashi
Department of Neurology and Clinical Neuroscience (H.N., M.O., M. Koga, M. Kawai, T.K.), Department of Laboratory Science (H.K.), Department of Pathology (E.I.), Yamaguchi University Graduate School of Medicine, Japan; Department of Neurology and Cerebrovascular Medicine (M.T.), Saitama International Medical Center, Saitama Medical University, Japan; and Department of Neurology and Geriatrics (Y.H., H.T.), Kagoshima University Graduate School of Medical and Dental Sciences, Japan.
Neurol Genet. 2017 Jul 27;3(4):e171. doi: 10.1212/NXG.0000000000000171. eCollection 2017 Aug.
To describe the autopsy case of a patient with a homozygous 2-base deletion, c171_172delGA (p.N58fs), in the gene.
We described the clinical history, neuroimaging data, neuropathology, and genetic analysis of the patients with mutations.
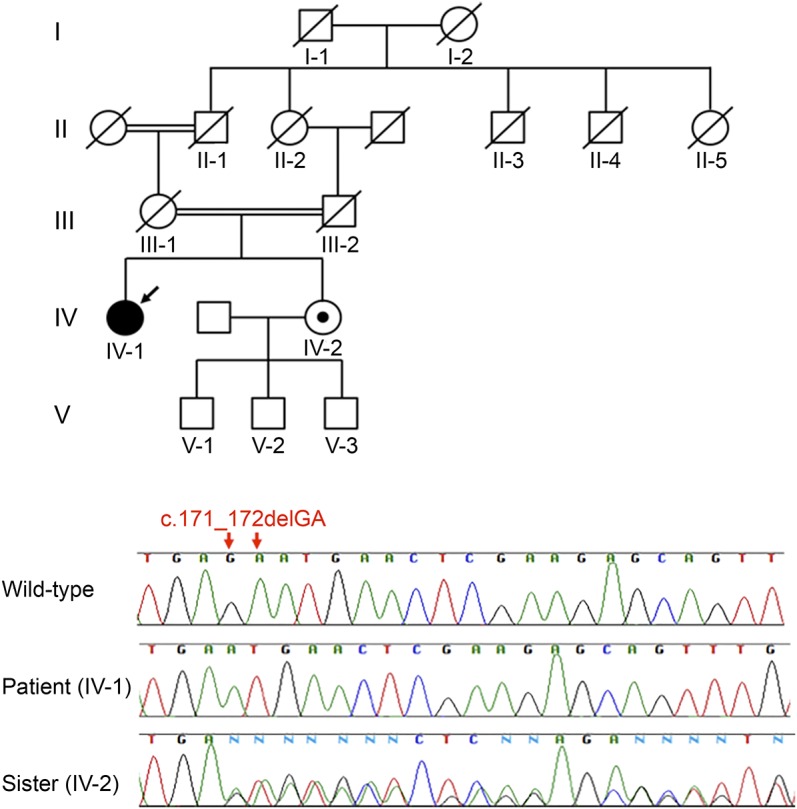
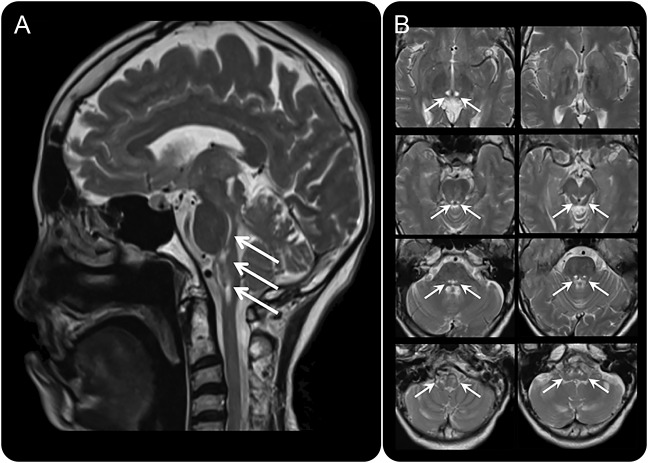
The patient was a Japanese woman with a history of delayed psychomotor development, primary amenorrhea, and gait disturbance in her 20s. She was hospitalized because of respiratory failure at the age of 60. Pectus excavatum, long fingers and toes, and pes cavus were revealed by physical examination. Her IQ score was 44. Neurologic examination revealed ophthalmoplegia, optic atrophy, dysphagia, distal dominant muscle weakness and atrophy, hyperreflexia at patellar tendon reflex, hyporeflexia at Achilles tendon reflex, and extensor plantar reflexes. At age 60, she died of pneumonia. Lactate levels were elevated in the patient's serum and CSF. T2-weighted brain MRI showed symmetrical hyperintense brainstem lesions. At autopsy, axial sections exposed symmetrical cyst formation with brownish lesions in the upper spinal cord, ventral medulla, pons, dorsal midbrain, and medial hypothalamus. Microscopic analysis of these areas demonstrated mild gliosis with rarefaction. Cell bodies in the choroid plexuses were eosinophilic and swollen. Electron microscopic examination revealed that these cells contained numerous abnormal mitochondria. Whole-exome sequencing revealed the 2-base deletion in .
We report an autopsy case of the mutation, and findings suggest that mitochondrial dysfunction may underlie the unique clinical presentations.
描述一名基因存在纯合2碱基缺失(c171_172delGA,p.N58fs)患者的尸检病例。
我们描述了该突变患者的临床病史、神经影像学数据、神经病理学和基因分析情况。
该患者为一名日本女性,有精神运动发育迟缓、原发性闭经病史,20多岁时出现步态障碍。她60岁时因呼吸衰竭住院。体格检查发现有漏斗胸、手指和脚趾细长以及高弓足。她的智商得分是44。神经系统检查发现有眼肌麻痹、视神经萎缩、吞咽困难、远端为主的肌肉无力和萎缩、髌腱反射亢进、跟腱反射减弱以及巴宾斯基征阳性。60岁时,她死于肺炎。患者血清和脑脊液中的乳酸水平升高。T2加权脑MRI显示脑干有对称性高信号病变。尸检时,轴位切片显示上脊髓、延髓腹侧、脑桥、中脑背侧和下丘脑内侧有对称性囊肿形成及褐色病变。对这些区域的显微镜分析显示有轻度胶质增生伴稀疏。脉络丛中的细胞体嗜酸性且肿胀。电子显微镜检查显示这些细胞含有大量异常线粒体。全外显子测序揭示了该基因中的2碱基缺失。
我们报告了一例该突变的尸检病例,研究结果表明线粒体功能障碍可能是其独特临床表现的基础。