The Dumont-UCLA Transplantation Center, Department of Surgery, Division of Liver and Pancreas Transplantation, David Geffen School of Medicine at University of California, Los Angeles, CA 90095, USA.
Department of Medicine, Division of Cardiology, David Geffen School of Medicine at University of California, Los Angeles, CA 90095, USA.
J Hepatol. 2017 Dec;67(6):1232-1242. doi: 10.1016/j.jhep.2017.08.010. Epub 2017 Aug 23.
BACKGROUND & AIMS: Hepatic ischemia-reperfusion injury (IRI), characterized by exogenous antigen-independent local inflammation and hepatocellular death, represents a risk factor for acute and chronic rejection in liver transplantation. We aimed to investigate the molecular communication involved in the mechanism of liver IRI.
We analyzed human liver transplants, primary murine macrophage cell cultures and IR-stressed livers in myeloid-specific heme oxygenase-1 (HO-1) gene mutant mice, for anti-inflammatory and cytoprotective functions of macrophage-specific HO-1/SIRT1 (sirtuin 1)/p53 (tumor suppressor protein) signaling.
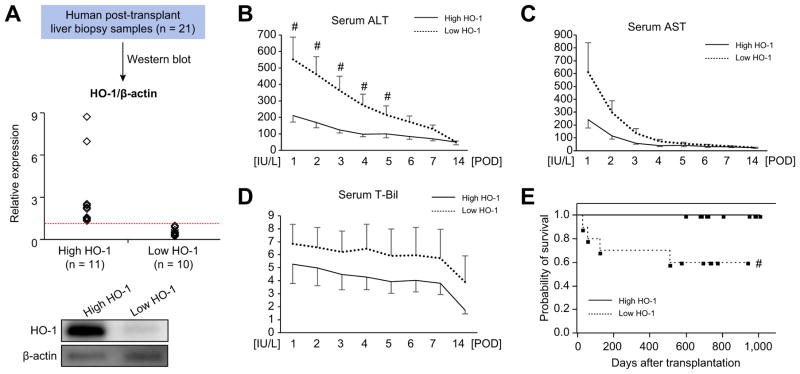
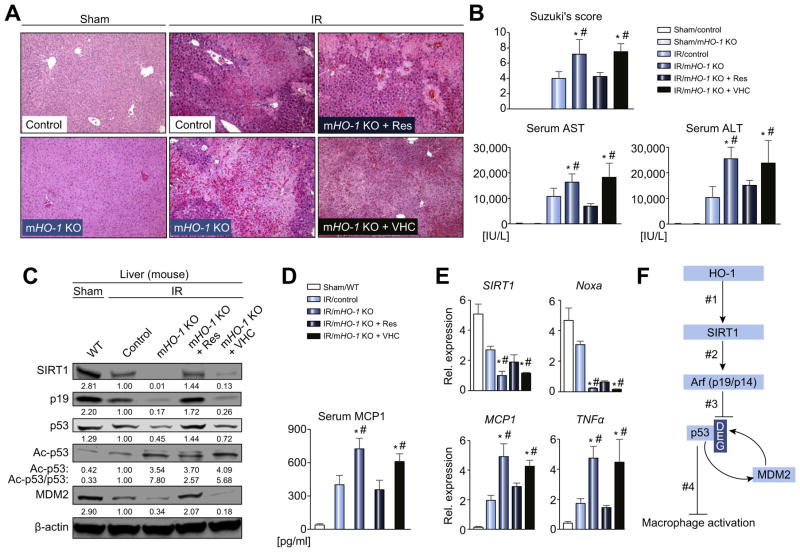
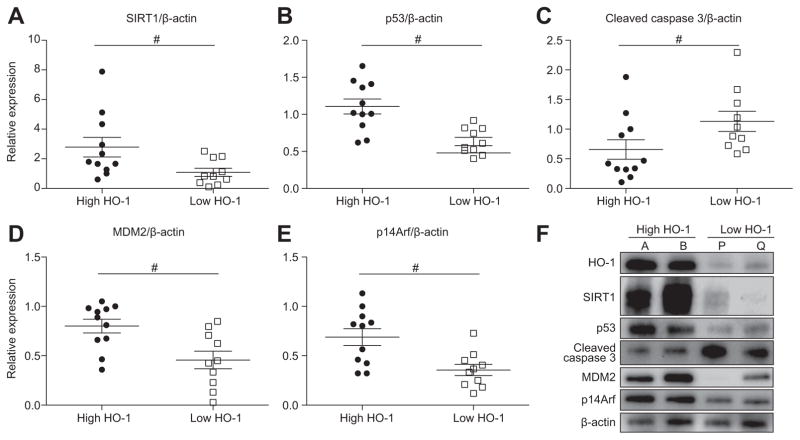
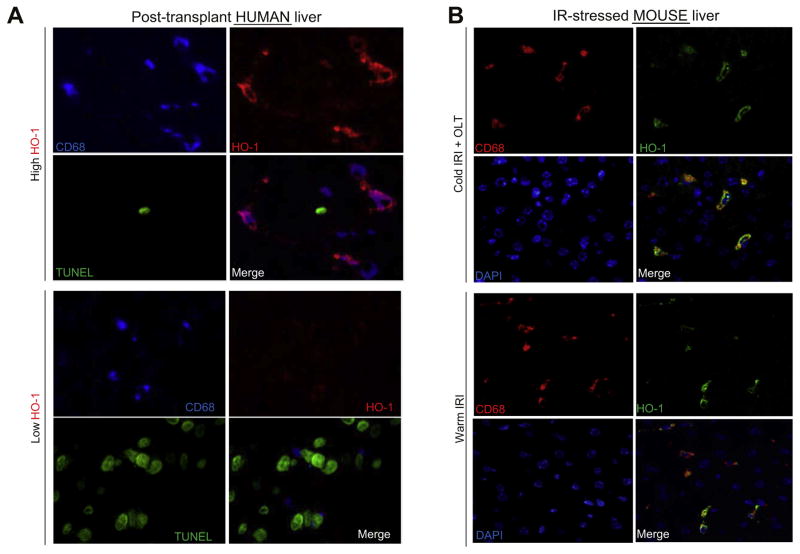
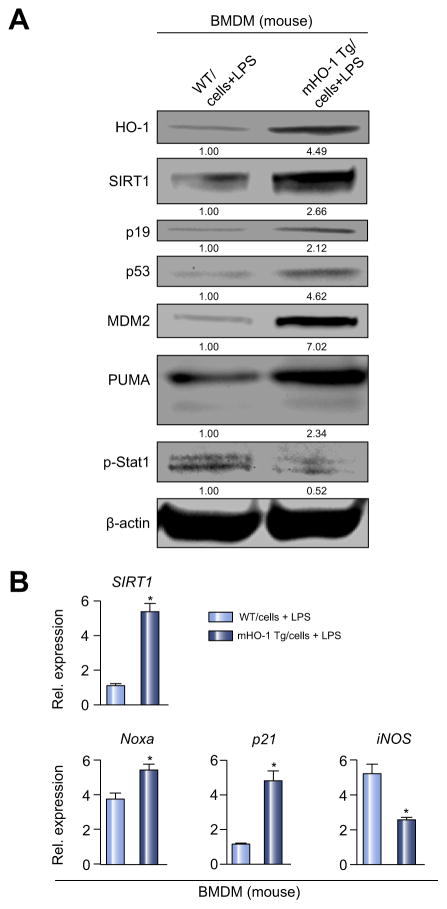
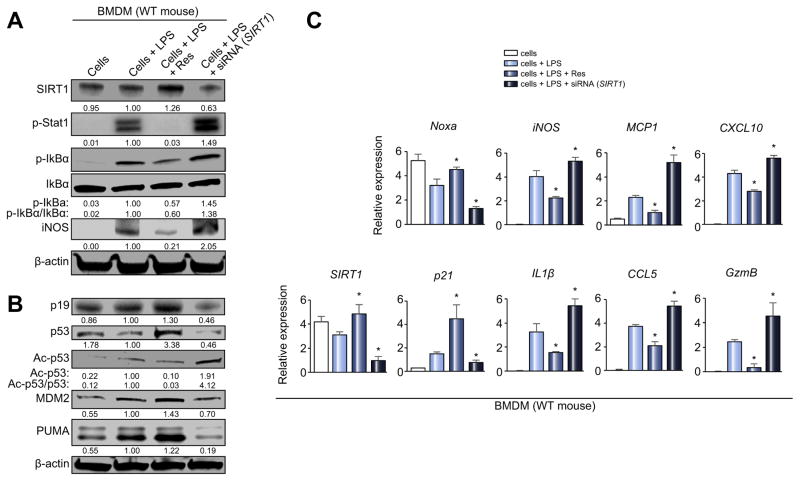
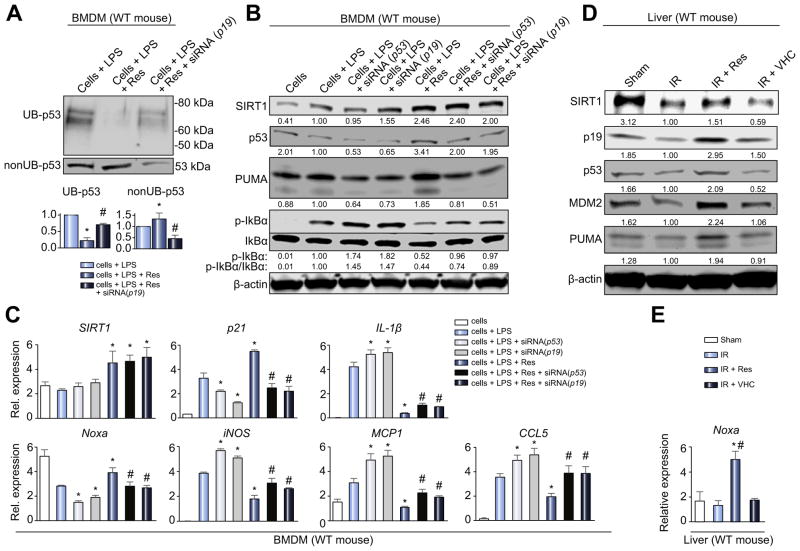
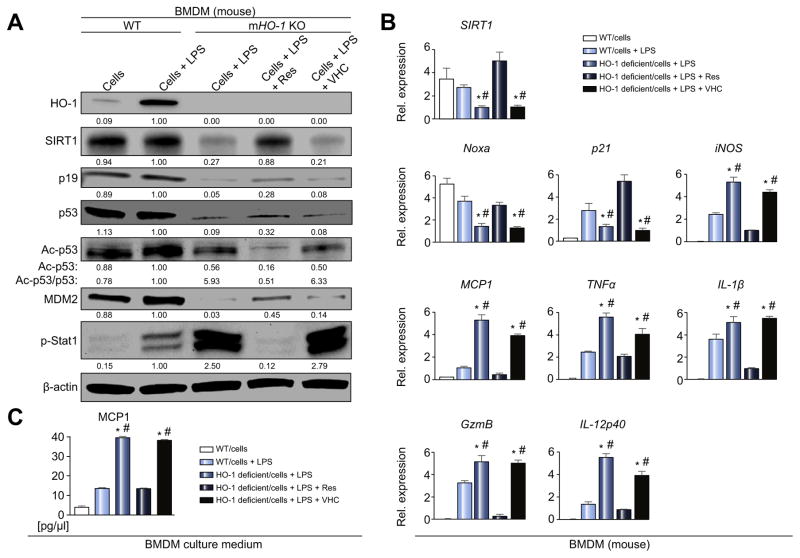
Decreased HO-1 expression in human post-reperfusion liver transplant biopsies correlated with a deterioration in hepatocellular function (serum ALT; p<0.05) and inferior patient survival (p<0.05). In the low HO-1 liver transplant biopsy group, SIRT1/Arf (alternative reading frame)/p53/MDM2 (murine double minute 2) expression levels decreased (p<0.05) while cleaved caspase 3 and frequency of TUNEL+cells simultaneously increased (p<0.05). Immunofluorescence showed macrophages were the principal source of HO-1 in human and mouse IR-stressed livers. In vitro macrophage cultures revealed that HO-1 induction positively regulated SIRT1 signaling, whereas SIRT1-induced Arf inhibited ubiquitinating activity of MDM2 against p53, which in turn attenuated macrophage activation. In a murine model of hepatic warm IRI, myeloid-specific HO-1 deletion lacked SIRT1/p53, exacerbated liver inflammation and IR-hepatocellular death, whereas adjunctive SIRT1 activation restored p53 signaling and rescued livers from IR-damage.
This bench-to-bedside study identifies a new class of macrophages activated via the HO-1-SIRT1-p53 signaling axis in the mechanism of hepatic sterile inflammation. This mechanism could be a target for novel therapeutic strategies in liver transplant recipients.
Post-transplant low macrophage HO-1 expression in human liver transplants correlates with reduced hepatocellular function and survival. HO-1 regulates macrophage activation via the SIRT1-p53 signaling network and regulates hepatocellular death in liver ischemia-reperfusion injury. Thus targeting this pathway in liver transplant recipients could be of therapeutic benefit.
肝脏缺血再灌注损伤(IRI)的特征为外源性抗原非依赖性局部炎症和肝细胞死亡,是肝移植中急性和慢性排斥的一个风险因素。本研究旨在探讨肝脏 IRI 机制中的分子通讯。
我们分析了人类肝移植、原代鼠巨噬细胞培养物和骨髓特异性血红素加氧酶-1(HO-1)基因突变小鼠的 IR 应激肝脏,以研究巨噬细胞特异性 HO-1/SIRT1(沉默调节蛋白 1)/p53(肿瘤抑制蛋白)信号通路的抗炎和细胞保护作用。
人再灌注肝移植活检中 HO-1 表达降低与肝细胞功能恶化(血清 ALT;p<0.05)和患者生存不良(p<0.05)相关。在低 HO-1 肝移植活检组中,SIRT1/Arf(替代阅读框)/p53/MDM2(鼠双微体 2)表达水平降低(p<0.05),而同时 cleaved caspase 3 和 TUNEL+细胞的频率增加(p<0.05)。免疫荧光显示巨噬细胞是人类和鼠 IR 应激肝脏中 HO-1 的主要来源。体外巨噬细胞培养显示,HO-1 诱导可正向调节 SIRT1 信号,而 SIRT1 诱导的 Arf 抑制 MDM2 对 p53 的泛素化活性,从而减弱巨噬细胞激活。在肝脏热 IRI 的鼠模型中,骨髓特异性 HO-1 缺失缺乏 SIRT1/p53,加重肝脏炎症和 IR 肝细胞死亡,而附加的 SIRT1 激活可恢复 p53 信号并使肝脏免受 IR 损伤。
这项从实验室到临床的研究在肝脏无菌炎症的机制中确定了一类新的通过 HO-1-SIRT1-p53 信号轴激活的巨噬细胞。该机制可能成为肝移植受者新的治疗策略的靶点。
人类肝移植中移植后巨噬细胞低 HO-1 表达与肝细胞功能降低和生存不良相关。HO-1 通过 SIRT1-p53 信号网络调节巨噬细胞激活,并调节肝脏缺血再灌注损伤中的肝细胞死亡。因此,在肝移植受者中靶向该途径可能具有治疗益处。