Gray Paul Edgar, Shadur Bella, Russell Susan, Mitchell Richard, Buckley Michael, Gallagher Kerri, Andrews Ian, Thia Kevin, Trapani Joseph A, Kirk Edwin Philip, Voskoboinik Ilia
Department of Immunology and Infectious Diseases, Sydney Children's Hospital, Randwick, NSW, Australia.
Kids Cancer Centre, Sydney Children's Hospital, Randwick, NSW, Australia.
Front Immunol. 2017 Aug 9;8:944. doi: 10.3389/fimmu.2017.00944. eCollection 2017.
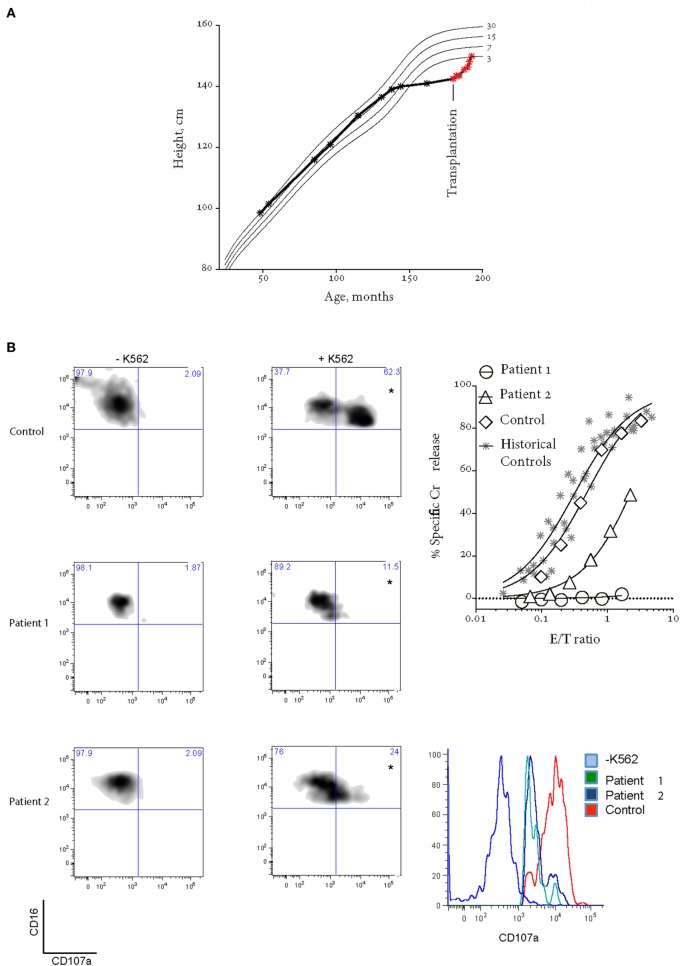
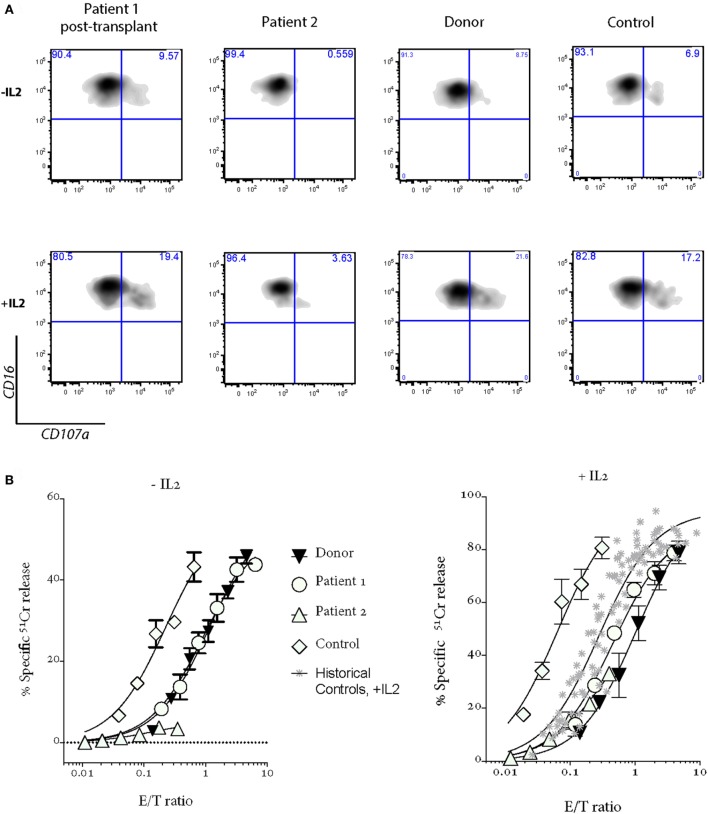
Bi-allelic null mutations affecting , or result in defects of lymphocyte cytotoxic degranulation and commonly cause familial hemophagocytic lymphohistiocytosis (FHL) in early life. Patients with partial loss of function are increasingly being diagnosed after presenting with alternative features of this disease, or with HLH later in life. Here, we studied two sisters with lymphocyte degranulation defects secondary to compound heterozygote missense variants in . The older sibling presented aged 11 with linear growth arrest and delayed puberty, 2 years prior to developing transient ischemic attacks secondary to neuroinflammation and hypogammaglobulinemia, but no FHL symptoms. Her geno-identical younger sister was initially asymptomatic but then presented at the same age with severe EBV-driven infectious mononucleosis, which was treated aggressively and did not progress to HLH. The sisters had similar natural killer cell degranulation; however, while cytotoxic activity was moderately reduced in the asymptomatic patient, it was completely absent in both siblings during active disease. Following allogeneic bone marrow transplantation at the age of 15, the older child has completely recovered NK cell cytotoxicity, is asymptomatic, and has experienced an exceptional compensatory growth spurt. Her younger sister was also successfully transplanted and is currently disease free. The current study reveals previously unappreciated manifestations of FHL in patients who inherited hypomorphic gene variants and also raises the important question of whether a threshold of minimum NK function can be defined that should protect a patient from serious disease manifestations such as HLH.
影响 或 的双等位基因无效突变会导致淋巴细胞细胞毒性脱颗粒缺陷,并通常在生命早期引起家族性噬血细胞性淋巴组织细胞增生症(FHL)。功能部分丧失的患者在出现该疾病的其他特征后,或在生命后期出现HLH时,越来越多地被诊断出来。在这里,我们研究了两名姐妹,她们因 基因的复合杂合子错义变异而继发淋巴细胞脱颗粒缺陷。姐姐11岁时出现线性生长停滞和青春期延迟,在因神经炎症和低丙种球蛋白血症继发短暂性脑缺血发作的2年前,未出现FHL症状。她基因相同的妹妹最初无症状,但在同一年龄出现严重的EB病毒驱动的传染性单核细胞增多症,经过积极治疗未进展为HLH。姐妹俩的自然杀伤细胞脱颗粒情况相似;然而,无症状患者的细胞毒性活性中度降低,而在疾病活动期,两姐妹的细胞毒性活性完全缺失。15岁时进行异基因骨髓移植后,年长的孩子NK细胞细胞毒性已完全恢复,无症状,并经历了一次异常的代偿性生长突增。她的妹妹也成功接受了移植,目前无病。本研究揭示了携带低功能基因变异的患者中FHL以前未被认识的表现,也提出了一个重要问题,即是否可以定义一个最低NK功能阈值,以保护患者免受HLH等严重疾病表现的影响。