Adhikari Badri, Cheng Jianlin
Department of Mathematics and Computer Science, University of Missouri-St.Louis, St. Louis, MO, 63121, USA.
Department of Electrical Engineering & Computer Science, Informatics Institute, University of Missouri, Columbia, MO, 65211, USA.
BMC Bioinformatics. 2017 Aug 29;18(1):380. doi: 10.1186/s12859-017-1807-5.
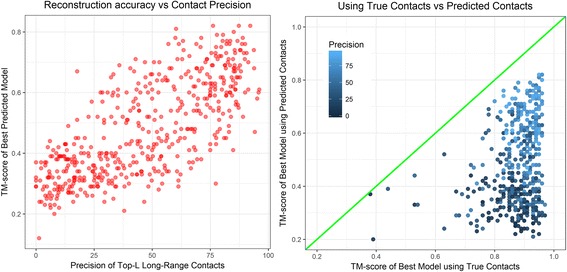
Residue-residue contacts are key features for accurate de novo protein structure prediction. For the optimal utilization of these predicted contacts in folding proteins accurately, it is important to study the challenges of reconstructing protein structures using true contacts. Because contact-guided protein modeling approach is valuable for predicting the folds of proteins that do not have structural templates, it is necessary for reconstruction studies to focus on hard-to-predict protein structures.
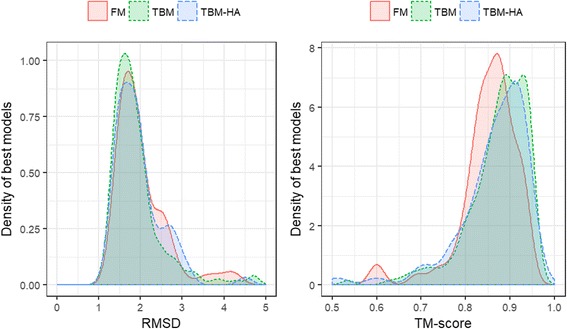
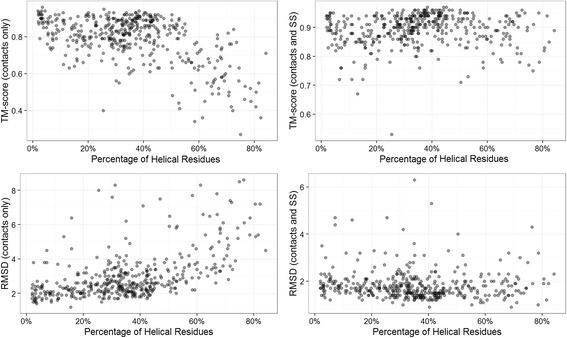
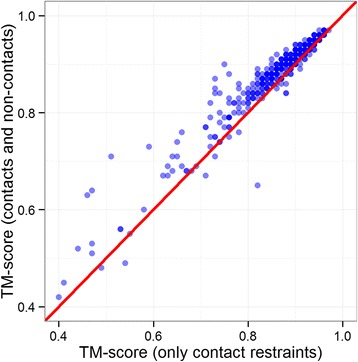
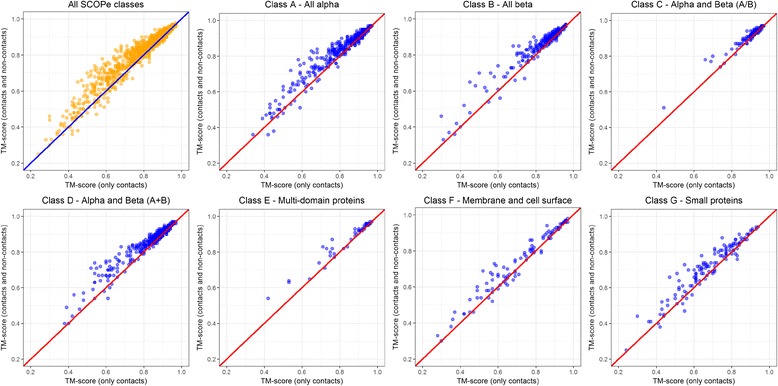
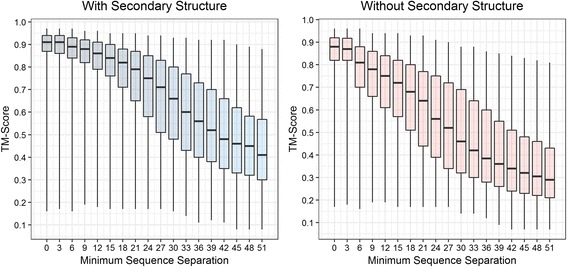
Using a data set consisting of 496 structural domains released in recent CASP experiments and a dataset of 150 representative protein structures, in this work, we discuss three techniques to improve the reconstruction accuracy using true contacts - adding secondary structures, increasing contact distance thresholds, and adding non-contacts. We find that reconstruction using secondary structures and contacts can deliver accuracy higher than using full contact maps. Similarly, we demonstrate that non-contacts can improve reconstruction accuracy not only when the used non-contacts are true but also when they are predicted. On the dataset consisting of 150 proteins, we find that by simply using low ranked predicted contacts as non-contacts and adding them as additional restraints, can increase the reconstruction accuracy by 5% when the reconstructed models are evaluated using TM-score.
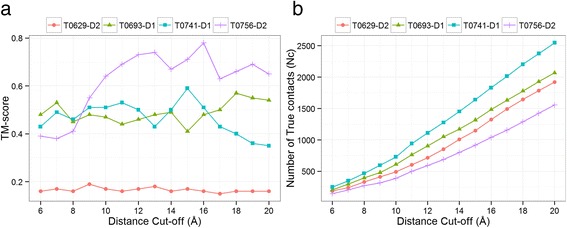
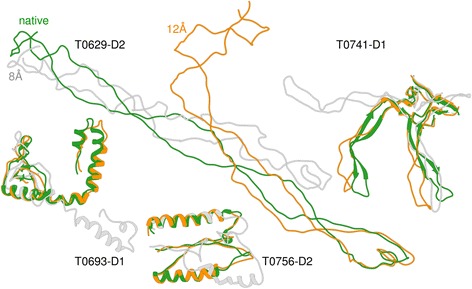
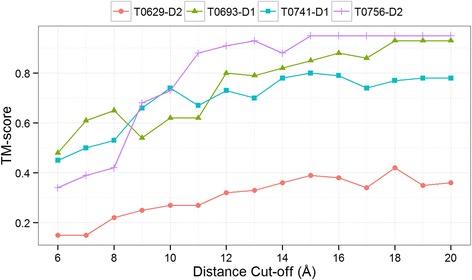
Our findings suggest that secondary structures are invaluable companions of contacts for accurate reconstruction. Confirming some earlier findings, we also find that larger distance thresholds are useful for folding many protein structures which cannot be folded using the standard definition of contacts. Our findings also suggest that for more accurate reconstruction using predicted contacts it is useful to predict contacts at higher distance thresholds (beyond 8 Å) and predict non-contacts.
残基-残基接触是准确进行蛋白质从头结构预测的关键特征。为了在准确折叠蛋白质时最佳利用这些预测接触,研究使用真实接触重建蛋白质结构所面临的挑战很重要。由于接触引导的蛋白质建模方法对于预测没有结构模板的蛋白质折叠很有价值,因此重建研究有必要聚焦于难以预测的蛋白质结构。
在这项工作中,我们使用由最近的蛋白质结构预测技术关键评估(CASP)实验中发布的496个结构域组成的数据集以及150个代表性蛋白质结构的数据集,讨论了三种使用真实接触提高重建准确性的技术——添加二级结构、增加接触距离阈值以及添加非接触。我们发现使用二级结构和接触进行重建可以提供比使用完整接触图更高的准确性。同样,我们证明非接触不仅在使用的非接触是真实的情况下,而且在它们是预测的情况下都可以提高重建准确性。在由150种蛋白质组成的数据集上,我们发现通过简单地将低排名的预测接触用作非接触并将它们作为额外的约束添加,当使用TM分数评估重建模型时,可以将重建准确性提高5%。
我们的研究结果表明,二级结构是准确重建中接触的宝贵辅助。证实了一些早期的发现,我们还发现更大的距离阈值对于折叠许多使用标准接触定义无法折叠的蛋白质结构很有用。我们的研究结果还表明,为了使用预测接触进行更准确的重建,在更高的距离阈值(超过8埃)预测接触并预测非接触是有用的。