Yao Ruen, Zhang Cheng, Yu Tingting, Li Niu, Hu Xuyun, Wang Xiumin, Wang Jian, Shen Yiping
Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Children's Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai, 200127 China.
Boston Children's Hospital, Boston, MA 02115 USA.
Mol Cytogenet. 2017 Aug 23;10:30. doi: 10.1186/s13039-017-0333-5. eCollection 2017.
Whole exome sequencing (WES) has been widely accepted as a robust and cost-effective approach for clinical genetic testing of small sequence variants. Detection of copy number variants (CNV) within WES data have become possible through the development of various algorithms and software programs that utilize read-depth as the main information. The aim of this study was to evaluate three commonly used, WES read-depth based CNV detection programs using high-resolution chromosomal microarray analysis (CMA) as a standard.
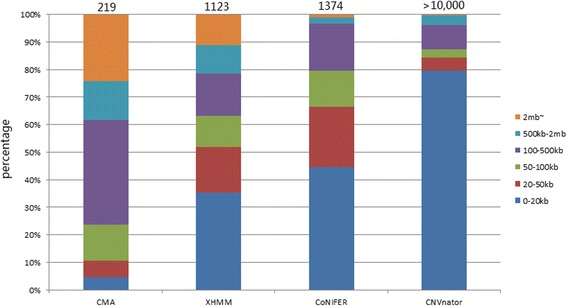
Paired CMA and WES data were acquired for 45 samples. A total of 219 CNVs (size ranged from 2.3 kb - 35 mb) identified on three CMA platforms (Affymetrix, Agilent and Illumina) were used as standards. CNVs were called from WES data using XHMM, CoNIFER, and CNVnator with modified settings.
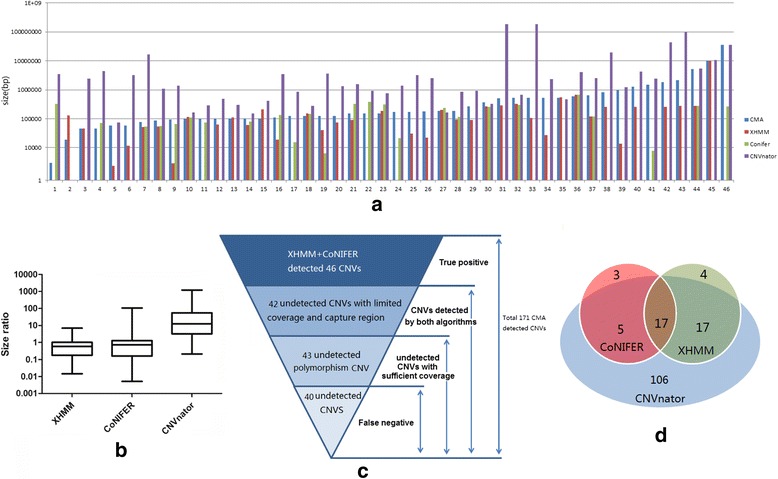
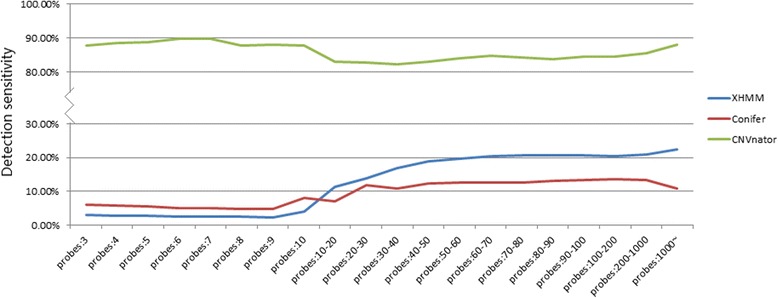
All three software packages detected an elevated proportion of small variants (< 20 kb) compared to CMA. XHMM and CoNIFER had poor detection sensitivity (22.2 and 14.6%), which correlated with the number of capturing probes involved. CNVnator detected most variants and had better sensitivity (87.7%); however, suffered from an overwhelming detection of small CNVs below 20 kb, which required further confirmation. Size estimation of variants was exaggerated by CNVnator and understated by XHMM and CoNIFER.
Low concordances of CNV, detected by three different read-depth based programs, indicate the immature status of WES-based CNV detection. Low sensitivity and uncertain specificity of WES-based CNV detection in comparison with CMA based CNV detection suggests that CMA will continue to play an important role in detecting clinical grade CNV in the NGS era, which is largely based on WES.
全外显子组测序(WES)作为一种用于小序列变异临床基因检测的强大且经济高效的方法已被广泛接受。通过开发各种利用读长深度作为主要信息的算法和软件程序,在WES数据中检测拷贝数变异(CNV)已成为可能。本研究的目的是以高分辨率染色体微阵列分析(CMA)为标准,评估三种常用的基于WES读长深度的CNV检测程序。
获取了45个样本的配对CMA和WES数据。在三个CMA平台(Affymetrix、Agilent和Illumina)上鉴定出的总共219个CNV(大小范围为2.3 kb - 35 mb)用作标准。使用经过修改设置的XHMM、CoNIFER和CNVnator从WES数据中调用CNV。
与CMA相比,所有这三个软件包检测到的小变异(<20 kb)比例都有所升高。XHMM和CoNIFER的检测灵敏度较差(分别为22.2%和14.6%),这与所涉及的捕获探针数量相关。CNVnator检测到的变异最多,灵敏度更高(87.7%);然而,它对低于20 kb的小CNV检测过多,需要进一步确认。CNVnator对变异大小的估计存在夸大,而XHMM和CoNIFER则低估了变异大小。
三种基于不同读长深度的程序检测到的CNV一致性较低,表明基于WES的CNV检测尚不成熟。与基于CMA的CNV检测相比,基于WES的CNV检测灵敏度低且特异性不确定,这表明在很大程度上基于WES的二代测序(NGS)时代,CMA将继续在检测临床级CNV中发挥重要作用。