Arber Charles, Angelova Plamena R, Wiethoff Sarah, Tsuchiya Yugo, Mazzacuva Francesca, Preza Elisavet, Bhatia Kailash P, Mills Kevin, Gout Ivan, Abramov Andrey Y, Hardy John, Duce James A, Houlden Henry, Wray Selina
Department of Molecular Neuroscience, Institute of Neurology, University College London, London, United Kingdom.
Institute of Structural and Molecular Biology, University College London, London, United Kingdom.
PLoS One. 2017 Sep 1;12(9):e0184104. doi: 10.1371/journal.pone.0184104. eCollection 2017.
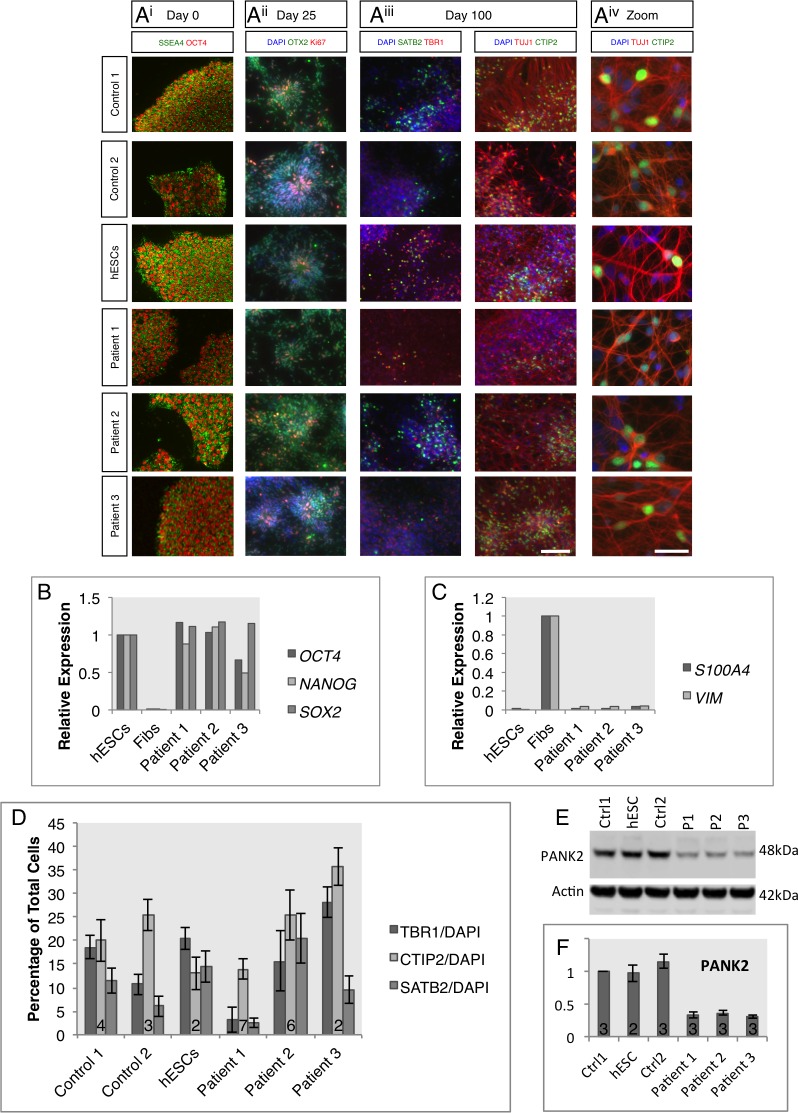
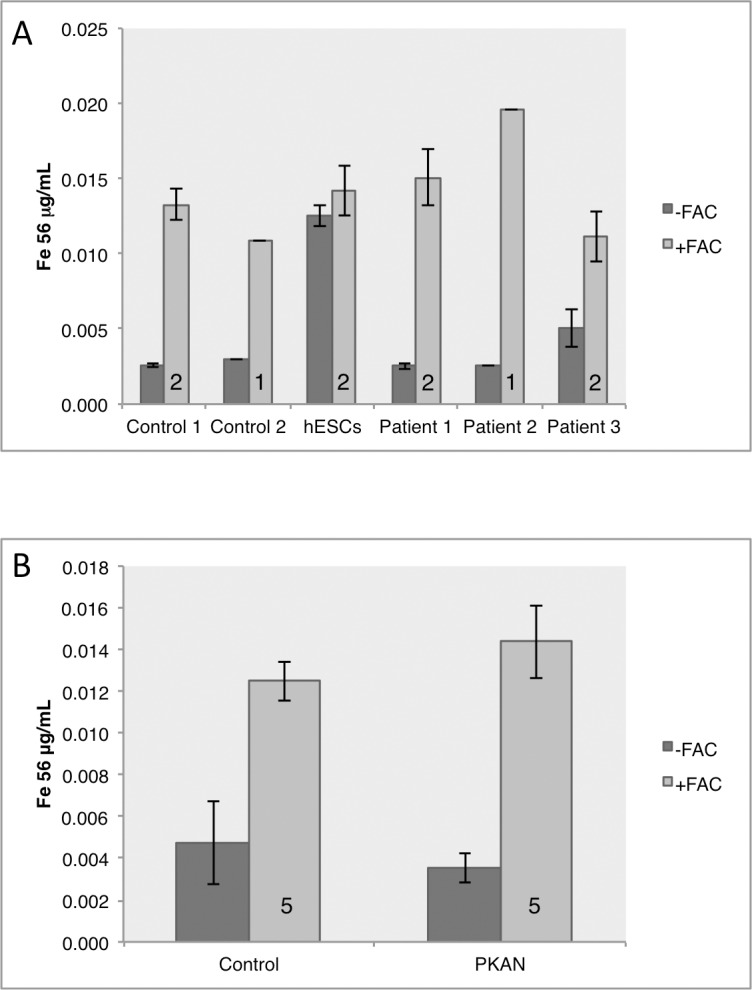
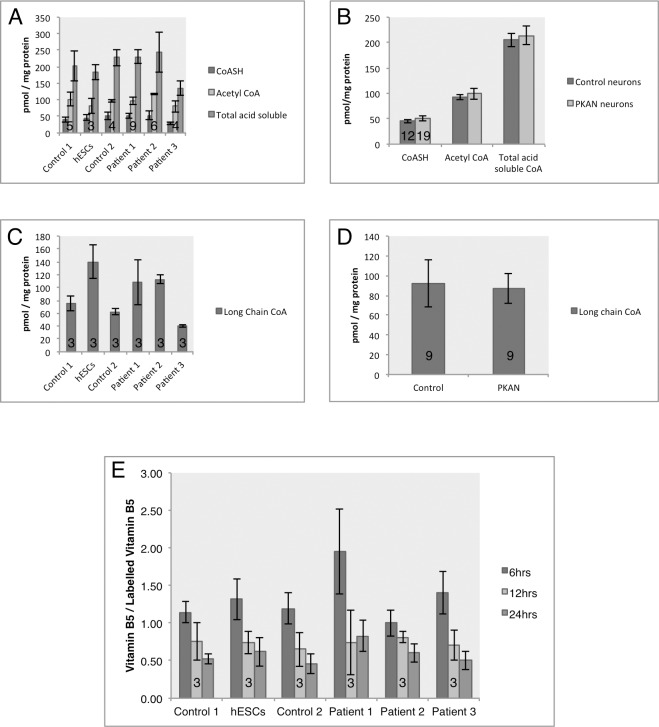
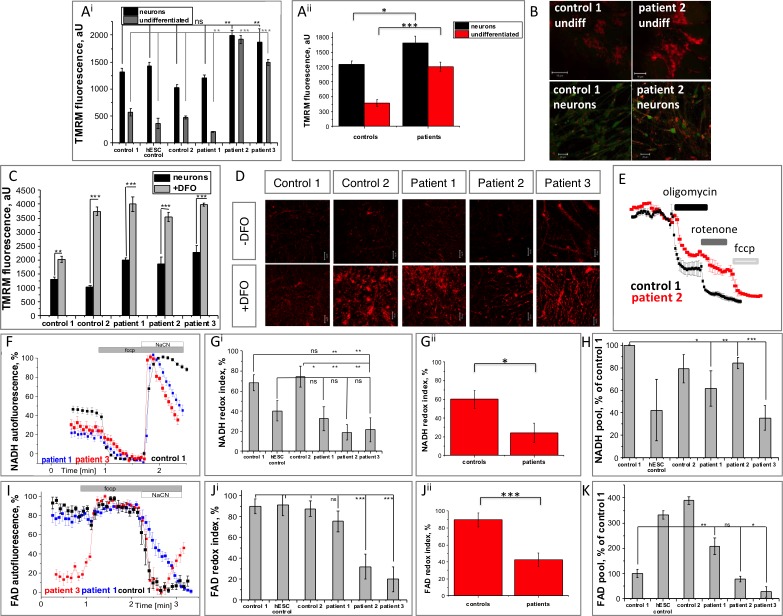
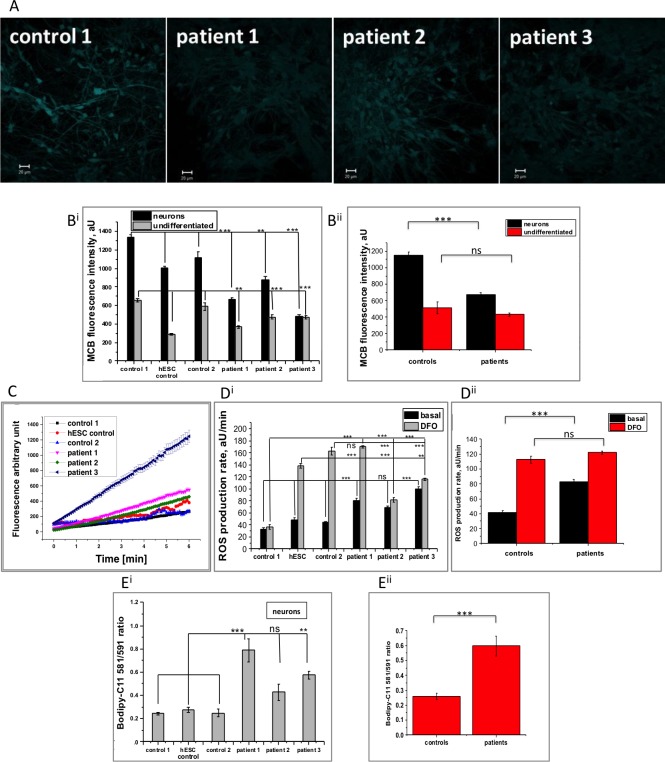
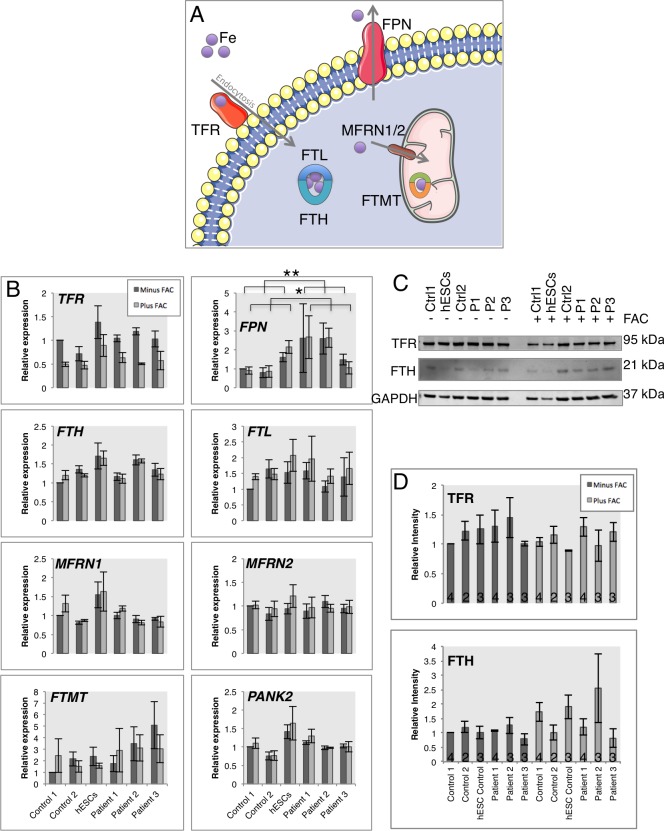
Mutations in PANK2 lead to neurodegeneration with brain iron accumulation. PANK2 has a role in the biosynthesis of coenzyme A (CoA) from dietary vitamin B5, but the neuropathological mechanism and reasons for iron accumulation remain unknown. In this study, atypical patient-derived fibroblasts were reprogrammed into induced pluripotent stem cells (iPSCs) and subsequently differentiated into cortical neuronal cells for studying disease mechanisms in human neurons. We observed no changes in PANK2 expression between control and patient cells, but a reduction in protein levels was apparent in patient cells. CoA homeostasis and cellular iron handling were normal, mitochondrial function was affected; displaying activated NADH-related and inhibited FADH-related respiration, resulting in increased mitochondrial membrane potential. This led to increased reactive oxygen species generation and lipid peroxidation in patient-derived neurons. These data suggest that mitochondrial deficiency is an early feature of the disease process and can be explained by altered NADH/FADH substrate supply to oxidative phosphorylation. Intriguingly, iron chelation appeared to exacerbate the mitochondrial phenotype in both control and patient neuronal cells. This raises caution for the use iron chelation therapy in general when iron accumulation is absent.
PANK2基因的突变会导致伴有脑铁蓄积的神经退行性变。PANK2在从膳食维生素B5生物合成辅酶A(CoA)的过程中发挥作用,但神经病理学机制以及铁蓄积的原因仍不清楚。在本研究中,将非典型患者来源的成纤维细胞重编程为诱导多能干细胞(iPSC),随后分化为皮质神经元细胞,以研究人类神经元中的疾病机制。我们观察到对照细胞和患者细胞之间PANK2表达没有变化,但患者细胞中蛋白质水平明显降低。CoA稳态和细胞铁处理正常,线粒体功能受到影响;表现为与NADH相关的呼吸激活和与FADH相关的呼吸抑制,导致线粒体膜电位升高。这导致患者来源的神经元中活性氧生成增加和脂质过氧化。这些数据表明线粒体缺陷是疾病过程的早期特征,并且可以通过氧化磷酸化中NADH/FADH底物供应的改变来解释。有趣的是,铁螯合似乎会加剧对照和患者神经元细胞中的线粒体表型。这对于在没有铁蓄积时普遍使用铁螯合疗法提出了警示。