School of Pharmacy, Ningxia Medical University, Yinchuan 750004, China.
Department of Pre-Clinical Sciences, Faculty of Medicine and Health Sciences, Universiti Tunku Abdul Rahman, Sungai Long campus, Kajang 43000, Selangor, Malaysia.
Molecules. 2017 Sep 22;22(10):1592. doi: 10.3390/molecules22101592.
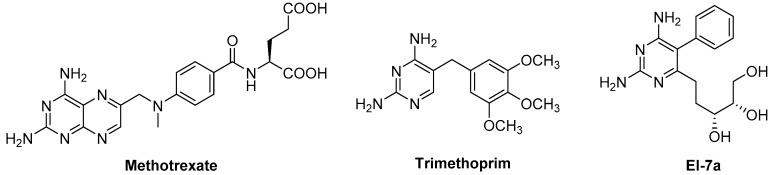
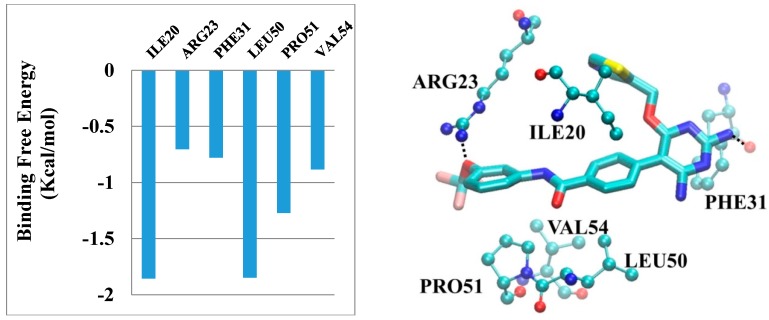
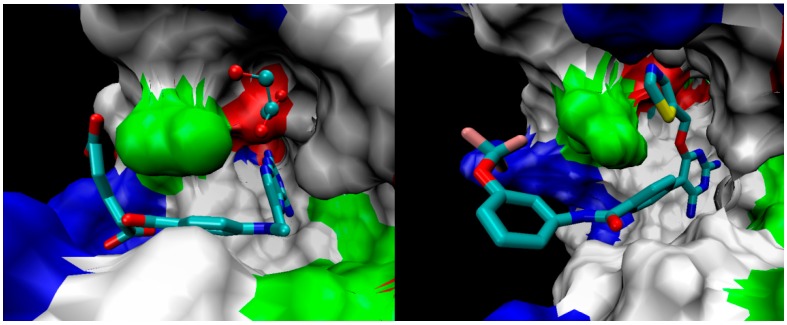
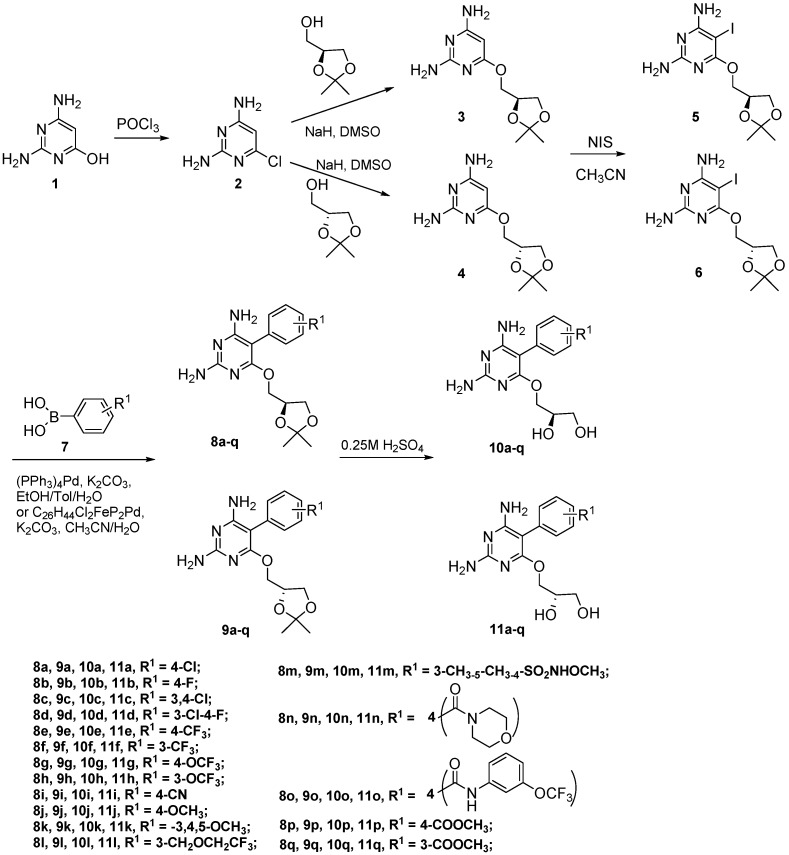
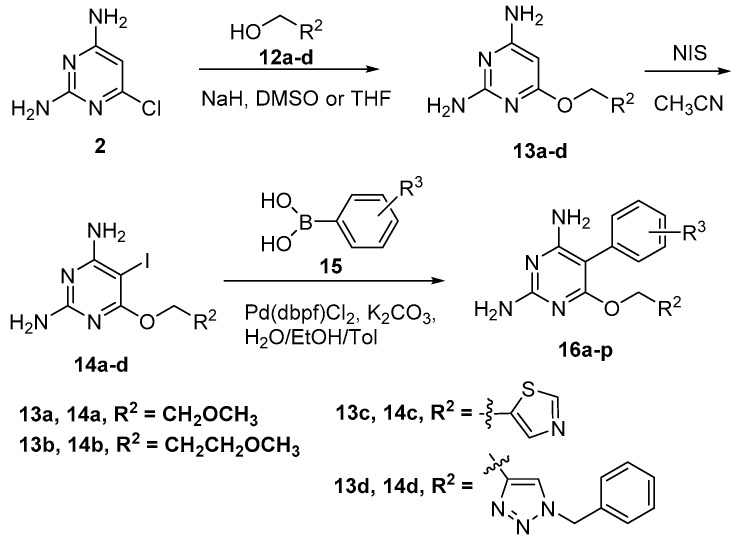
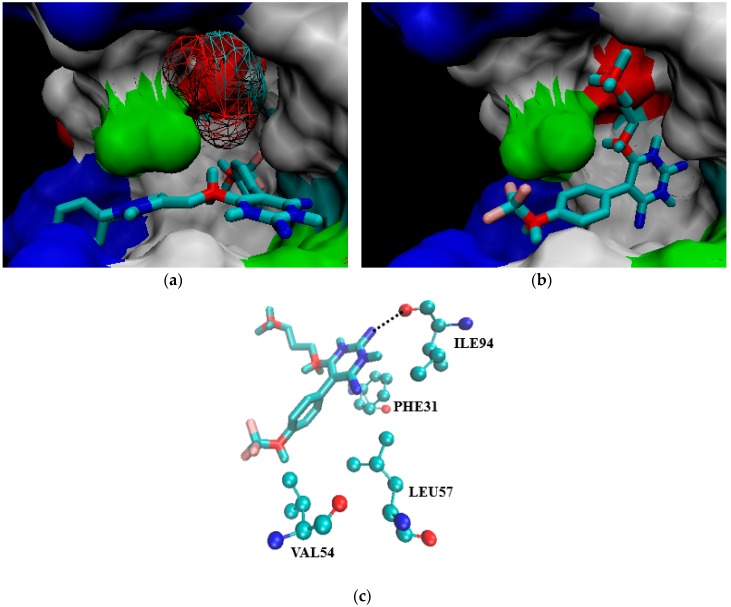
Tuberculosis (TB) is a chronic, potentially fatal disease caused by (). The dihyrofolate reductase in (-DHFR) is believed to be an important drug target in anti-TB drug development. This enzyme contains a glycerol (GOL) binding site, which is assumed to be a useful site to improve the selectivity towards human dihyrofolate reductase (-DHFR). There have been previous attempts to design drugs targeting the GOL binding site, but the designed compounds contain a hydrophilic group, which may prevent the compounds from crossing the cell wall of to function at the whole cell level. In the current study, we designed and synthesized a series of -DHFR inhibitors that contain a 2,4-diaminopyrimidine core with side chains to occupy the glycerol binding site with proper hydrophilicity for cell entry, and tested their anti-tubercular activity against H37Ra. Among them, compound showed a good anti-TB activity (MIC = 6.25 μg/mL) with a significant selectivity against vero cells. In the molecular simulations performed to understand the binding poses of the compounds, it was noticed that only side chains of a certain size can occupy the glycerol binding site. In summary, the novel synthesized compounds with appropriate side chains, hydrophobicity and selectivity could be important lead compounds for future optimization towards the development of future anti-TB drugs that can be used as monotherapy or in combination with other anti-TB drugs or antibiotics. These compounds can also provide much information for further studies on -DHFR. However, the enzyme target of the compounds still needs to be confirmed by pure -DHFR binding assays.
结核病(TB)是一种由()引起的慢性、潜在致命疾病。二氢叶酸还原酶(DHFR)被认为是抗结核药物开发中的一个重要药物靶点。该酶含有甘油(GOL)结合位点,该位点被认为是提高对人二氢叶酸还原酶(DHFR)选择性的有用位点。之前曾有人试图设计针对 GOL 结合位点的药物,但设计的化合物含有亲水性基团,这可能会阻止化合物穿过细胞壁,从而在整个细胞水平上发挥作用。在目前的研究中,我们设计并合成了一系列含有 2,4-二氨基嘧啶核心并带有侧链的 DHFR 抑制剂,这些侧链可以占据甘油结合位点,并具有适当的亲水性以进入细胞,我们测试了它们对 H37Ra 的抗结核活性。其中,化合物 表现出良好的抗结核活性(MIC = 6.25 μg/mL),对 vero 细胞具有显著的选择性。在进行的分子模拟中,我们注意到只有一定大小的侧链才能占据甘油结合位点。综上所述,具有适当侧链、疏水性和选择性的新型合成化合物可能是未来优化抗结核药物开发的重要先导化合物,可单独或与其他抗结核药物或抗生素联合使用。这些化合物还可以为进一步研究 DHFR 提供更多信息。然而,化合物的酶靶标仍需要通过纯 DHFR 结合测定来确认。