Feichtinger René G, Oláhová Monika, Kishita Yoshihito, Garone Caterina, Kremer Laura S, Yagi Mikako, Uchiumi Takeshi, Jourdain Alexis A, Thompson Kyle, D'Souza Aaron R, Kopajtich Robert, Alston Charlotte L, Koch Johannes, Sperl Wolfgang, Mastantuono Elisa, Strom Tim M, Wortmann Saskia B, Meitinger Thomas, Pierre Germaine, Chinnery Patrick F, Chrzanowska-Lightowlers Zofia M, Lightowlers Robert N, DiMauro Salvatore, Calvo Sarah E, Mootha Vamsi K, Moggio Maurizio, Sciacco Monica, Comi Giacomo P, Ronchi Dario, Murayama Kei, Ohtake Akira, Rebelo-Guiomar Pedro, Kohda Masakazu, Kang Dongchon, Mayr Johannes A, Taylor Robert W, Okazaki Yasushi, Minczuk Michal, Prokisch Holger
Department of Pediatrics, University Hospital Salzburg, Paracelsus Medical University, 5020 Salzburg, Austria.
Wellcome Centre for Mitochondrial Research, Institute of Neuroscience and Institute for Cell and Molecular Biosciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK.
Am J Hum Genet. 2017 Oct 5;101(4):525-538. doi: 10.1016/j.ajhg.2017.08.015. Epub 2017 Sep 21.
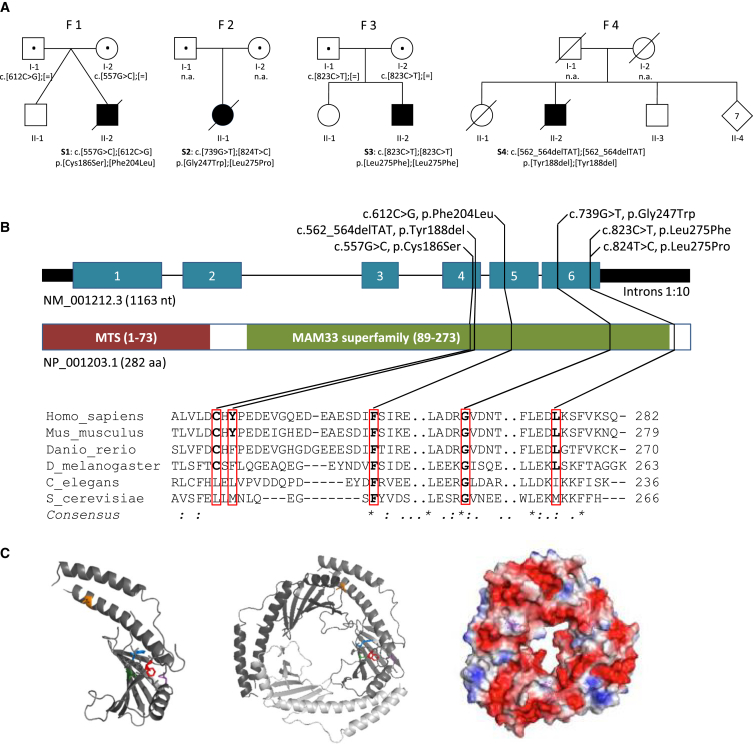
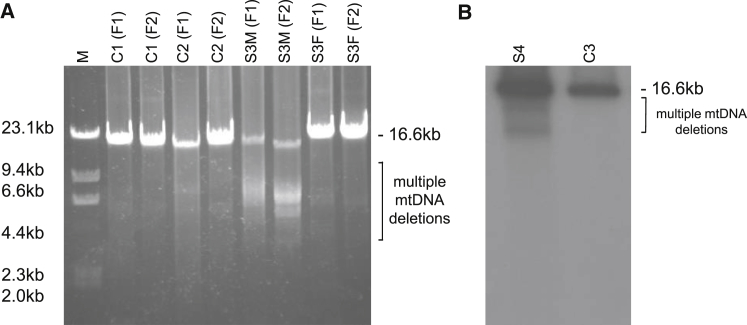
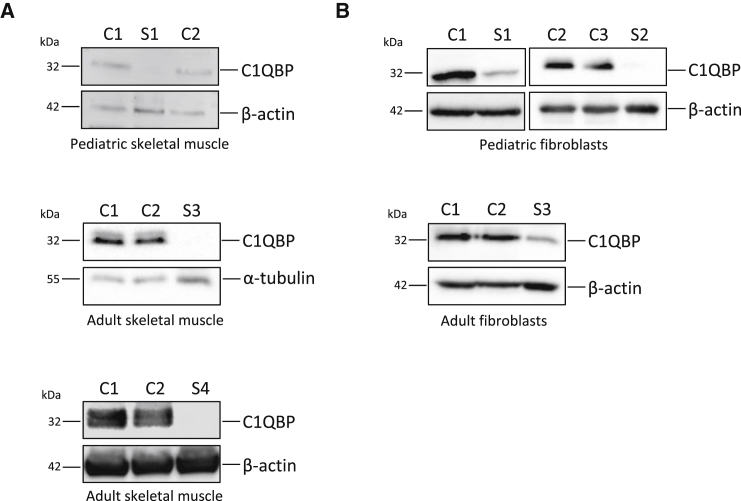
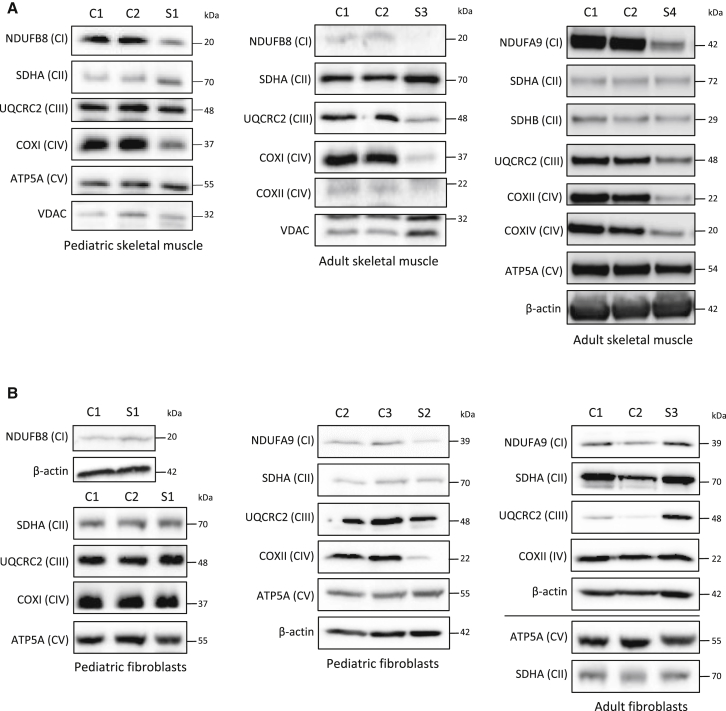
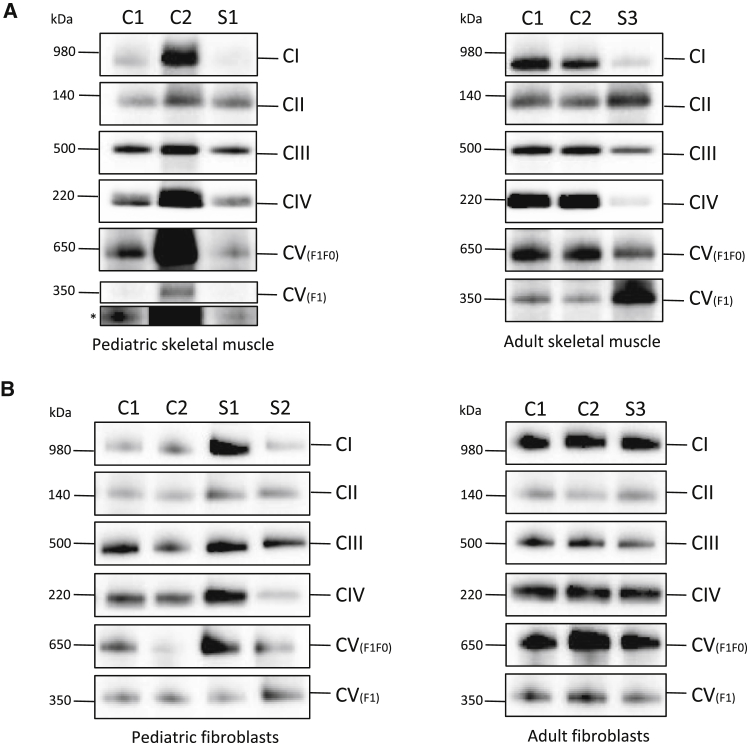
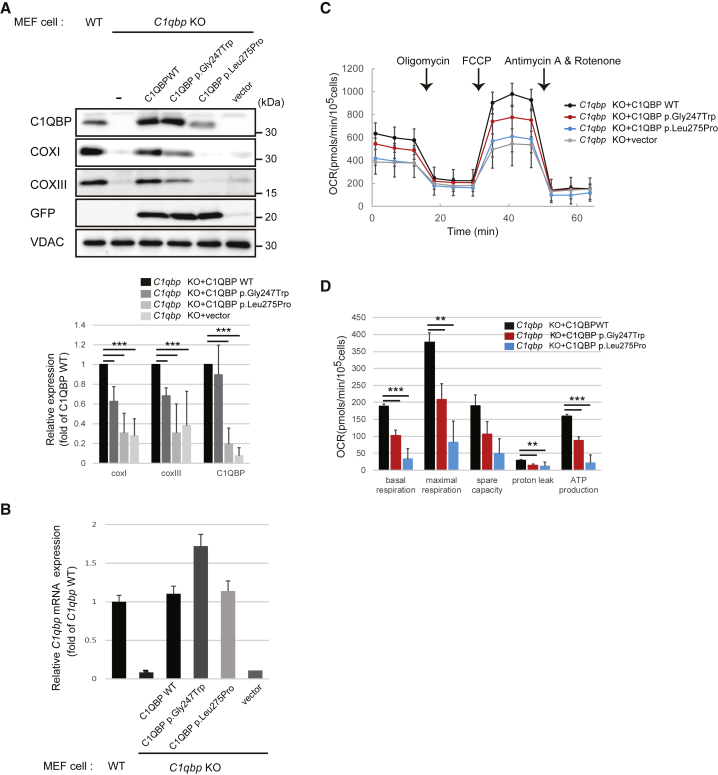
Complement component 1 Q subcomponent-binding protein (C1QBP; also known as p32) is a multi-compartmental protein whose precise function remains unknown. It is an evolutionary conserved multifunctional protein localized primarily in the mitochondrial matrix and has roles in inflammation and infection processes, mitochondrial ribosome biogenesis, and regulation of apoptosis and nuclear transcription. It has an N-terminal mitochondrial targeting peptide that is proteolytically processed after import into the mitochondrial matrix, where it forms a homotrimeric complex organized in a doughnut-shaped structure. Although C1QBP has been reported to exert pleiotropic effects on many cellular processes, we report here four individuals from unrelated families where biallelic mutations in C1QBP cause a defect in mitochondrial energy metabolism. Infants presented with cardiomyopathy accompanied by multisystemic involvement (liver, kidney, and brain), and children and adults presented with myopathy and progressive external ophthalmoplegia. Multiple mitochondrial respiratory-chain defects, associated with the accumulation of multiple deletions of mitochondrial DNA in the later-onset myopathic cases, were identified in all affected individuals. Steady-state C1QBP levels were decreased in all individuals' samples, leading to combined respiratory-chain enzyme deficiency of complexes I, III, and IV. C1qbp mouse embryonic fibroblasts (MEFs) resembled the human disease phenotype by showing multiple defects in oxidative phosphorylation (OXPHOS). Complementation with wild-type, but not mutagenized, C1qbp restored OXPHOS protein levels and mitochondrial enzyme activities in C1qbp MEFs. C1QBP deficiency represents an important mitochondrial disorder associated with a clinical spectrum ranging from infantile lactic acidosis to childhood (cardio)myopathy and late-onset progressive external ophthalmoplegia.
补体成分1Q亚成分结合蛋白(C1QBP;也称为p32)是一种多区室蛋白,其确切功能尚不清楚。它是一种进化保守的多功能蛋白,主要定位于线粒体基质,在炎症和感染过程、线粒体核糖体生物发生以及细胞凋亡和核转录的调节中发挥作用。它有一个N端线粒体靶向肽,在导入线粒体基质后被蛋白水解加工,在那里它形成一个以甜甜圈形状组织的同三聚体复合物。尽管据报道C1QBP对许多细胞过程具有多效性作用,但我们在此报告了来自无关家族的4名个体,其中C1QBP的双等位基因突变导致线粒体能量代谢缺陷。婴儿表现为心肌病并伴有多系统受累(肝脏、肾脏和大脑),儿童和成人表现为肌病和进行性眼外肌麻痹。在所有受影响个体中均发现了多个线粒体呼吸链缺陷,这与迟发性肌病病例中线粒体DNA的多个缺失的积累有关。所有个体样本中的稳态C1QBP水平均降低,导致复合物I、III和IV的呼吸链酶联合缺乏。C1qbp小鼠胚胎成纤维细胞(MEFs)通过显示氧化磷酸化(OXPHOS)中的多个缺陷而类似于人类疾病表型。用野生型而非诱变的C1qbp进行互补可恢复C1qbp MEFs中的OXPHOS蛋白水平和线粒体酶活性。C1QBP缺乏代表一种重要的线粒体疾病,其临床谱范围从婴儿乳酸酸中毒到儿童(心脏)肌病和迟发性进行性眼外肌麻痹。