Kitakaze Keisuke, Tasaki Chikako, Tajima Youichi, Hirokawa Takatsugu, Tsuji Daisuke, Sakuraba Hitoshi, Itoh Kohji
Department of Medicinal Biotechnology, Institute of Biomedical Sciences, Graduate School of Pharmaceutical Science, Tokushima University, 1-78-1, Tokushima 770-8505, Japan.
Molecular Medical Research Project, Department of Genome Medicine, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan.
Biochem Biophys Rep. 2016 Jun 8;7:157-163. doi: 10.1016/j.bbrep.2016.04.012. eCollection 2016 Sep.
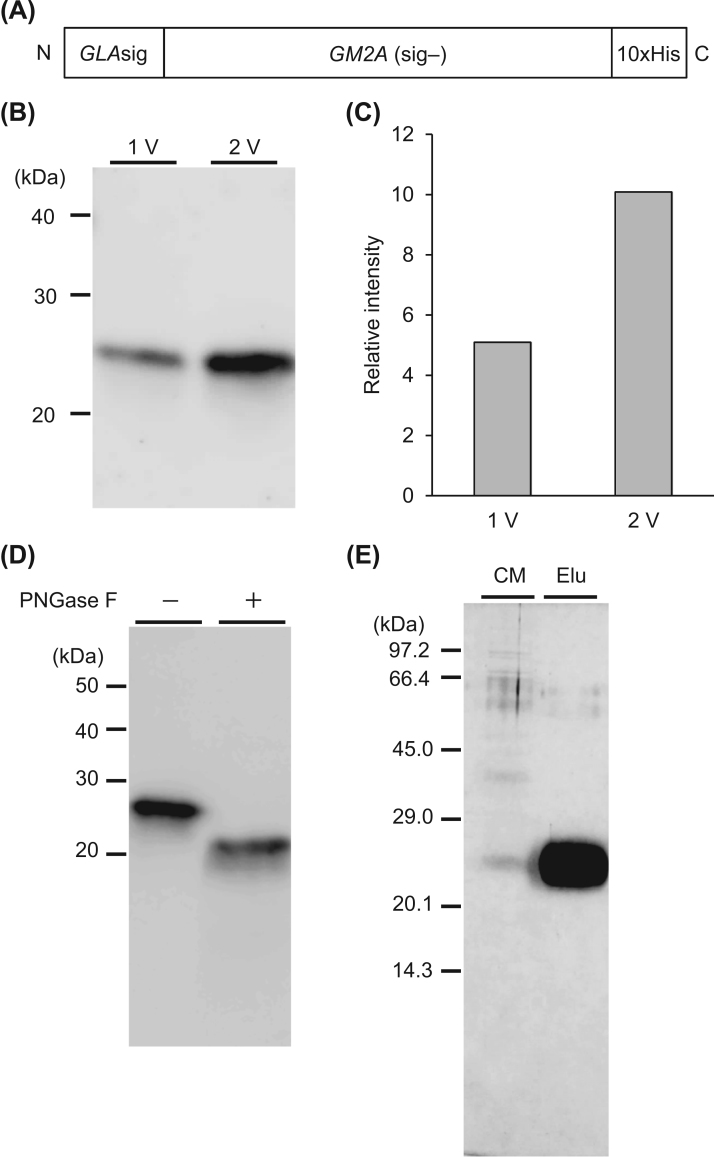
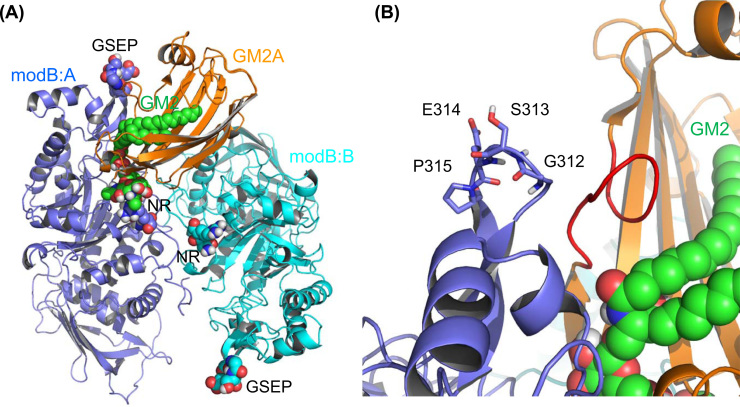
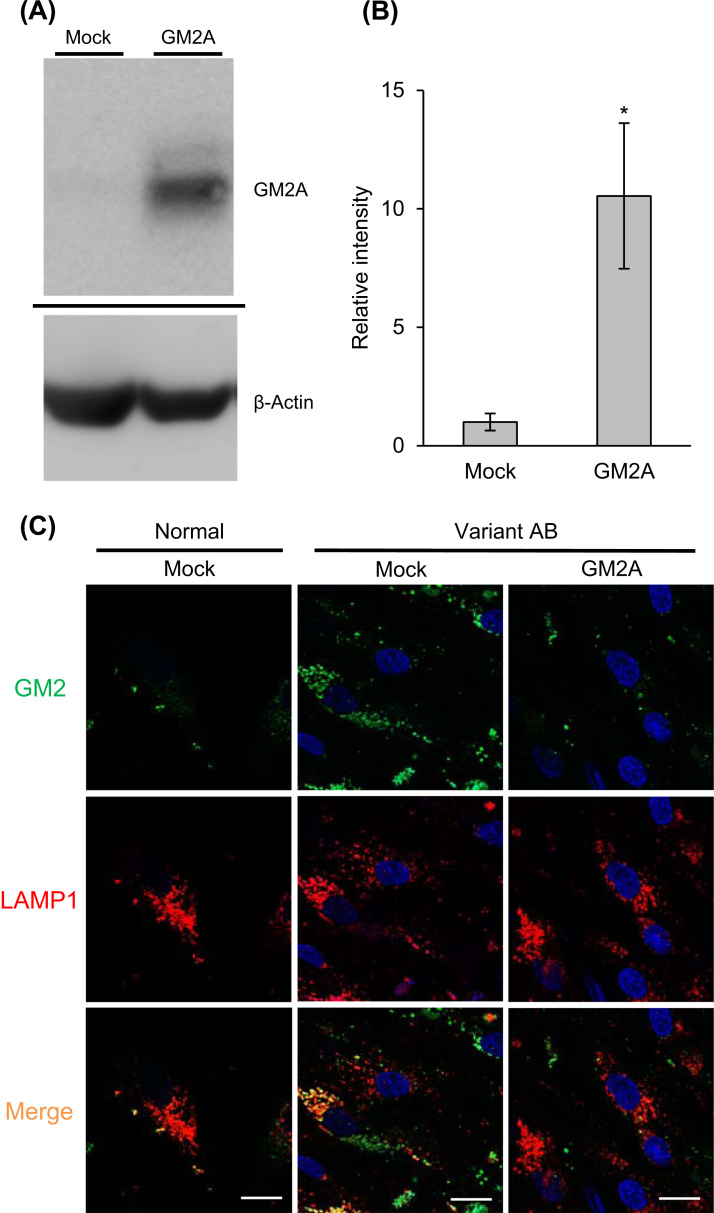
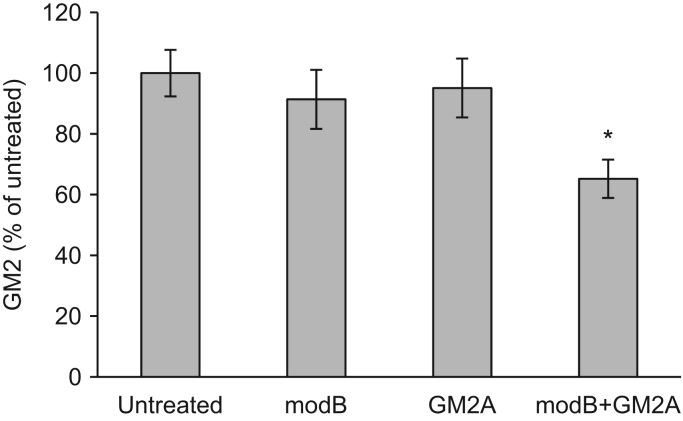
GM2 gangliosidoses are autosomal recessive lysosomal storage diseases (LSDs) caused by mutations in the , and genes, which encode the human lysosomal β-hexosaminidase (Hex) α- and β-subunits, and GM2 activator protein (GM2A), respectively. These diseases are associated with excessive accumulation of GM2 ganglioside (GM2) in the brains of patients with neurological symptoms. Here we established a CHO cell line overexpressing human and purified GM2A from the conditioned medium, which was taken up by fibroblasts derived from a patient with GM2A deficiency, and had the therapeutic effects of reducing the GM2 accumulated in fibroblasts when added to the culture medium. We also demonstrated for the first time that recombinant GM2A could enhance the replacement effect of human modified HexB (modB) with GM2-degrading activity, which is composed of homodimeric altered β-subunits containing a partial amino acid sequence of the α-subunit, including the GSEP loop necessary for binding to GM2A, on reduction of the GM2 accumulated in fibroblasts derived from a patient with Tay-Sachs disease, a HexA (αβ heterodimer) deficiency, caused by mutations. We predicted the same manner of binding of GM2A to the GSEP loop located in the modified HexB β-subunit to that in the native HexA α-subunit on the basis of the x-ray crystal structures. These findings suggest the effectiveness of combinational replacement therapy involving the human modified HexB and GM2A for GM2 gangliosidoses.
GM2神经节苷脂沉积症是常染色体隐性溶酶体贮积病(LSDs),由、和基因的突变引起,这些基因分别编码人类溶酶体β - 己糖胺酶(Hex)的α和β亚基以及GM2激活蛋白(GM2A)。这些疾病与患有神经症状患者大脑中GM2神经节苷脂(GM2)的过度积累有关。在此,我们建立了一个过表达人类的CHO细胞系,并从条件培养基中纯化了GM2A,GM2A被来自一名GM2A缺乏症患者的成纤维细胞摄取,当添加到培养基中时,具有减少成纤维细胞中积累的GM2的治疗效果。我们还首次证明,重组GM2A可以增强具有GM2降解活性的人类修饰HexB(modB)的替代作用,modB由含有α亚基部分氨基酸序列的同型二聚体改变的β亚基组成,包括与GM2A结合所需的GSEP环,可减少由突变引起的泰 - 萨克斯病(一种HexA(αβ异二聚体)缺乏症)患者来源的成纤维细胞中积累的GM2。基于X射线晶体结构,我们预测GM2A与修饰HexBβ亚基中GSEP环的结合方式与天然HexAα亚基中的相同。这些发现表明,涉及人类修饰HexB和GM2A的联合替代疗法对GM2神经节苷脂沉积症有效。