Drug Transporter Sciences, Cyprotex Discovery Ltd (an Evotec company), No 24 Mereside, Alderley Park, Macclesfield, Cheshire, United Kingdom.
Pharmacol Res Perspect. 2017 Oct;5(5). doi: 10.1002/prp2.357.
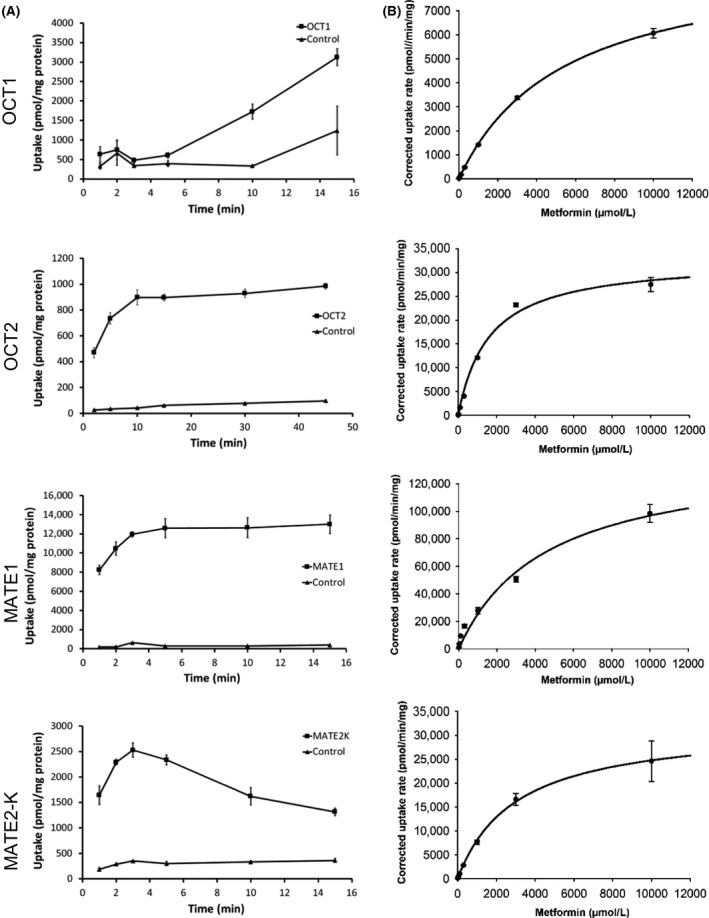
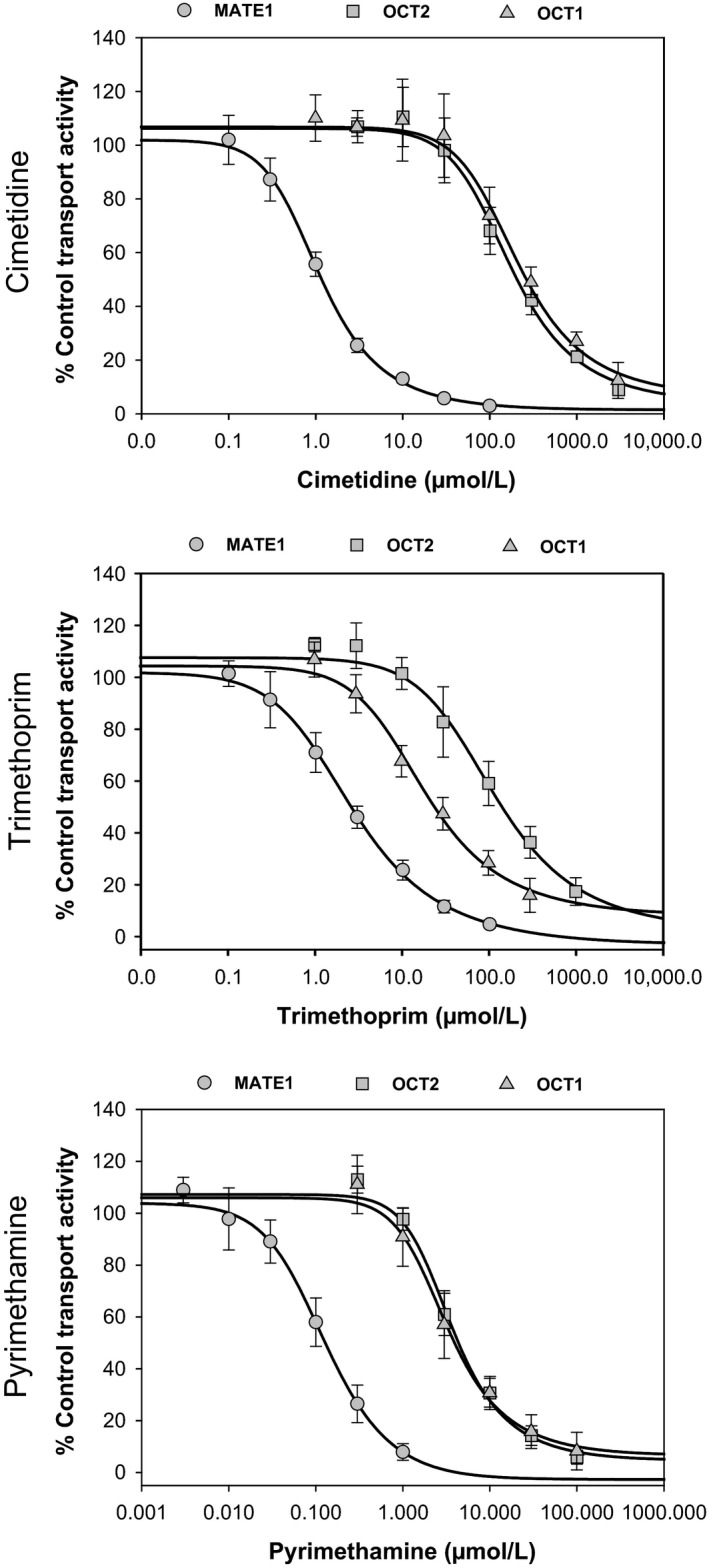
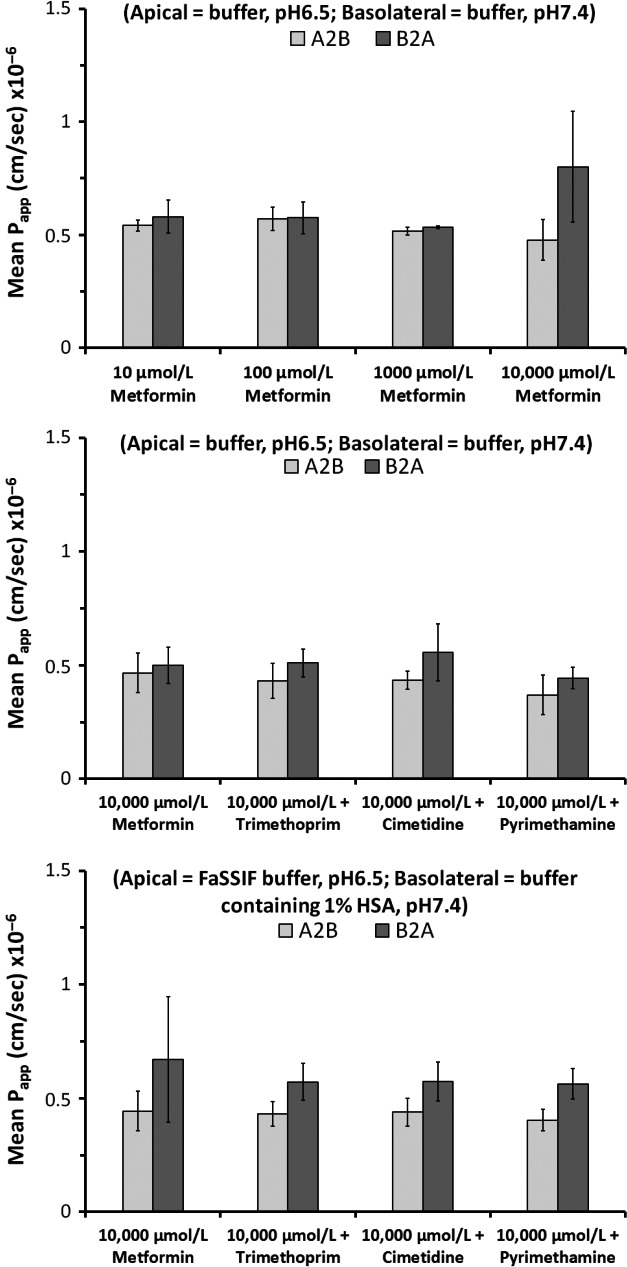
Metformin is a common co-medication for many diseases and the victim of clinical drug-drug interactions (DDIs) perpetrated by cimetidine, trimethoprim and pyrimethamine, resulting in decreased active renal clearance due to inhibition of organic cation transport proteins and increased plasma exposure of metformin. To understand whether area under the plasma concentration-time curve (AUC) increases relate to absorption, in vitro inhibitory potencies of these drugs against metformin transport by human organic cation transporter (OCT) 1, and the apical to basolateral absorptive permeability of metformin across Caco-2 cells in the presence of therapeutic intestinal concentrations of cimetidine, trimethoprim or pyrimethamine, were determined. Whilst all inhibited OCT1, none enhanced metformin's absorptive permeability (~0.5 × 10 cm/sec) suggesting that DDI AUC changes are not related to absorption. Subsequently, to understand whether inhibition of renal transporters are responsible for AUC increases, in vitro inhibitory potencies against metformin transport by human OCT2, multidrug and toxin extrusion (MATE) 1 and MATE2-K were determined. Ensuing IC values were incorporated into mechanistic static equations, alongside unbound maximal plasma concentration and transporter fraction excreted values, in order to calculate theoretical increases in metformin AUC due to inhibition by cimetidine, trimethoprim or pyrimethamine. Calculated theoretical fold-increases in metformin exposure confirmed solitary inhibition of renal MATE1 to be the likely mechanism underlying the observed exposure changes in clinical DDIs. Interestingly, clinically observed increases in metformin AUC were predicted more closely when the renal transporter fraction excreted value derived from oral metformin administration, rather than intravenous, was utilized in theoretical calculations, likely reflecting the "flip-flop" pharmacokinetic profile of the drug.
二甲双胍是许多疾病的常用合并用药,也是西咪替丁、甲氧苄啶和乙胺嘧啶等临床药物相互作用(DDI)的受害者,导致有机阳离子转运蛋白抑制导致活性肾清除率降低,以及二甲双胍的血浆暴露增加。为了了解 AUC 增加是否与吸收有关,测定了这些药物对人有机阳离子转运蛋白(OCT)1 转运二甲双胍的体外抑制效力,以及在西咪替丁、甲氧苄啶或乙胺嘧啶的治疗肠道浓度存在下,二甲双胍在 Caco-2 细胞中的顶端至基底侧吸收渗透性。虽然所有药物都抑制了 OCT1,但没有一种药物增强了二甲双胍的吸收渗透性(~0.5×10-5cm/sec),这表明 DDI AUC 变化与吸收无关。随后,为了了解抑制肾脏转运体是否是 AUC 增加的原因,测定了这些药物对人 OCT2、多药和毒素外排(MATE)1 和 MATE2-K 转运二甲双胍的体外抑制效力。随后将 IC 值纳入到机制静态方程中,同时考虑未结合的最大血浆浓度和转运体排泄分数值,以计算由于西咪替丁、甲氧苄啶或乙胺嘧啶抑制导致二甲双胍 AUC 的理论增加。计算出的二甲双胍暴露理论增加倍数证实,肾脏 MATE1 的单独抑制可能是临床 DDI 中观察到的暴露变化的潜在机制。有趣的是,当在理论计算中使用从口服二甲双胍给药中获得的肾脏转运体排泄分数值,而不是静脉内给药时,更密切地预测了临床观察到的二甲双胍 AUC 增加,这可能反映了药物的“翻转”药代动力学特征。