Mesbah-Uddin Md, Guldbrandtsen Bernt, Iso-Touru Terhi, Vilkki Johanna, De Koning Dirk-Jan, Boichard Didier, Lund Mogens Sandø, Sahana Goutam
Department of Molecular Biology and Genetics, Center for Quantitative Genetics and Genomics, Aarhus University, 8830 Tjele, Denmark.
Animal Genetics and Integrative Biology, UMR 1313 GABI, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France.
DNA Res. 2018 Feb;25(1):49-59. doi: 10.1093/dnares/dsx037. Epub 2017 Sep 8.
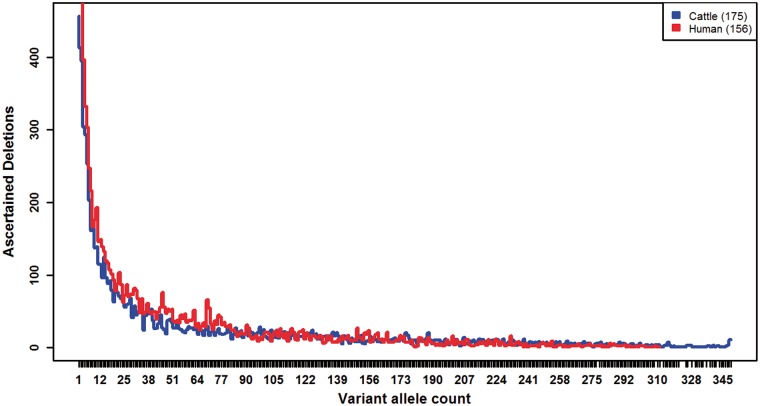
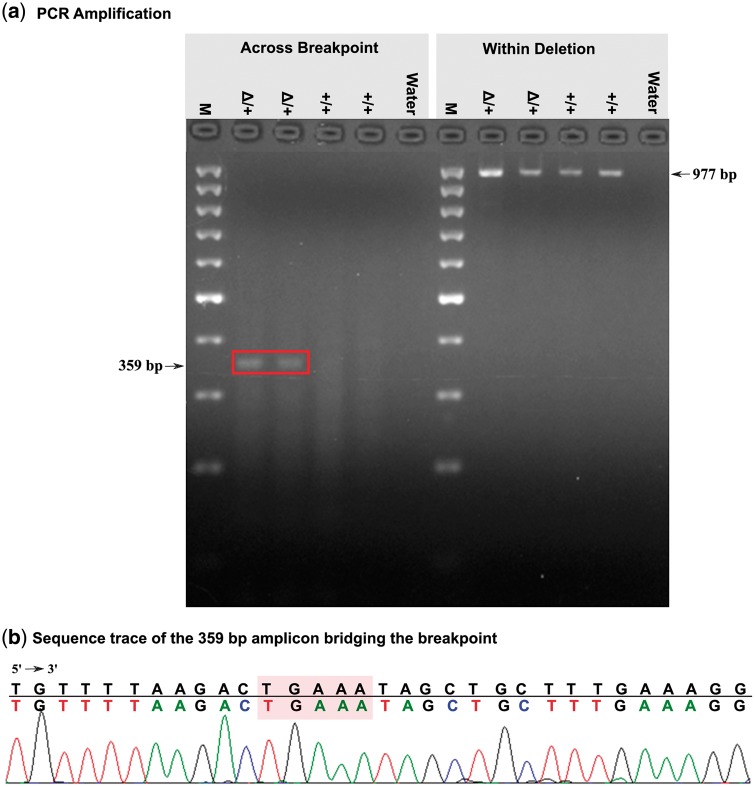
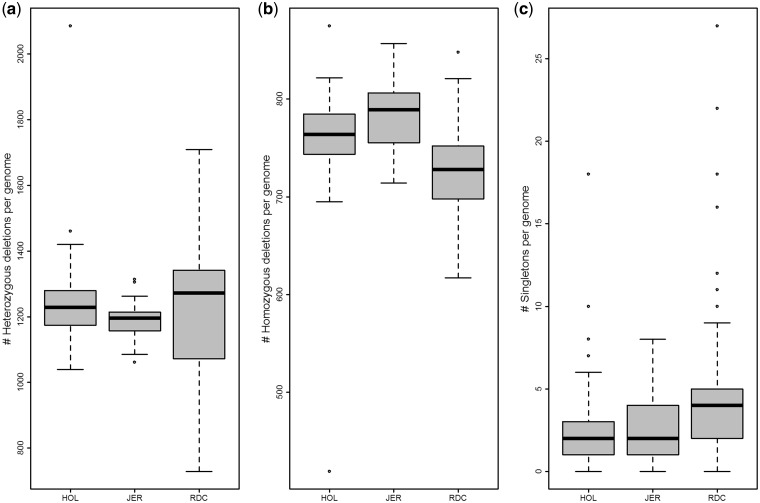
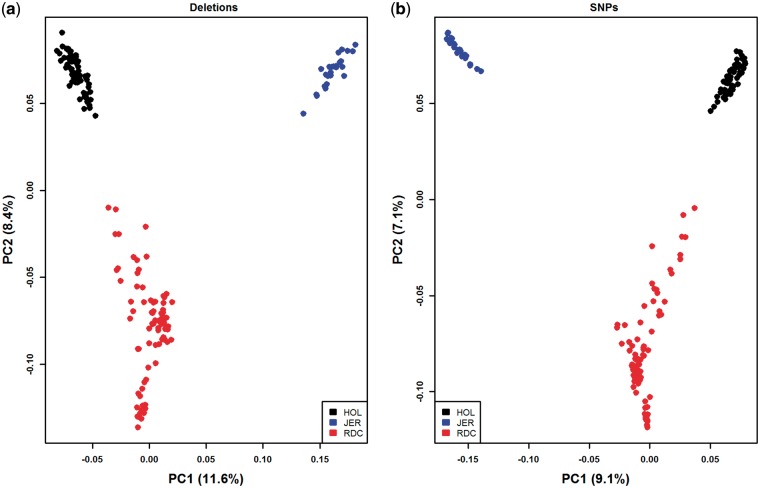
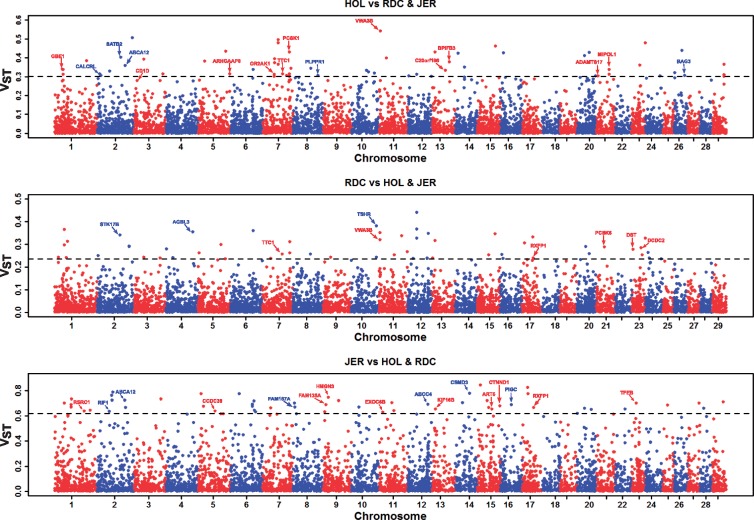
Large genomic deletions are potential candidate for loss-of-function, which could be lethal as homozygote. Analysing whole genome data of 175 cattle, we report 8,480 large deletions (199 bp-773 KB) with an overall false discovery rate of 8.8%; 82% of which are novel compared with deletions in the dbVar database. Breakpoint sequence analyses revealed that majority (24 of 29 tested) of the deletions contain microhomology/homology at breakpoint, and therefore, most likely generated by microhomology-mediated end joining. We observed higher differentiation among breeds for deletions in some genic-regions, such as ABCA12, TTC1, VWA3B, TSHR, DST/BPAG1, and CD1D. The genes overlapping deletions are on average evolutionarily less conserved compared with known mouse lethal genes (P-value = 2.3 × 10-6). We report 167 natural gene knockouts in cattle that are apparently nonessential as live homozygote individuals are observed. These genes are functionally enriched for immunoglobulin domains, olfactory receptors, and MHC classes (FDR = 2.06 × 10-22, 2.06 × 10-22, 7.01 × 10-6, respectively). We also demonstrate that deletions are enriched for health and fertility related quantitative trait loci (2-and 1.5-fold enrichment, Fisher's P-value = 8.91 × 10-10 and 7.4 × 10-11, respectively). Finally, we identified and confirmed the breakpoint of a ∼525 KB deletion on Chr23:12,291,761-12,817,087 (overlapping BTBD9, GLO1 and DNAH8), causing stillbirth in Nordic Red Cattle.
大型基因组缺失是功能丧失的潜在候选因素,作为纯合子可能是致死性的。通过分析175头牛的全基因组数据,我们报告了8480个大型缺失(199bp - 773KB),总体错误发现率为8.8%;与dbVar数据库中的缺失相比,其中82%是新发现的。断点序列分析表明,大多数(29个测试中有24个)缺失在断点处含有微同源性/同源性,因此很可能是由微同源性介导的末端连接产生的。我们观察到在一些基因区域(如ABCA12、TTC1、VWA3B、TSHR、DST/BPAG1和CD1D)的缺失在品种间具有更高的分化。与已知的小鼠致死基因相比,重叠缺失的基因在进化上平均保守性较低(P值 = 2.3×10 - 6)。我们报告了牛中有167个自然基因敲除,显然作为纯合子活体个体是无害的。这些基因在功能上富集于免疫球蛋白结构域、嗅觉受体和MHC类别(FDR分别为2.06×10 - 22、2.06×10 - 22、7.01×10 - 6)。我们还证明,缺失在与健康和生育相关的数量性状位点上富集(分别富集2倍和1.5倍,Fisher's P值 = 8.91×10 - 10和7.4×10 - 11)。最后,我们鉴定并确认了Chr23:12,291,761 - 12,817,087上一个约525KB缺失(重叠BTBD9、GLO1和DNAH8)的断点,该缺失导致北欧红牛死产。