Department of Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115, USA; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA.
Cell Rep. 2017 Oct 10;21(2):467-481. doi: 10.1016/j.celrep.2017.09.056.
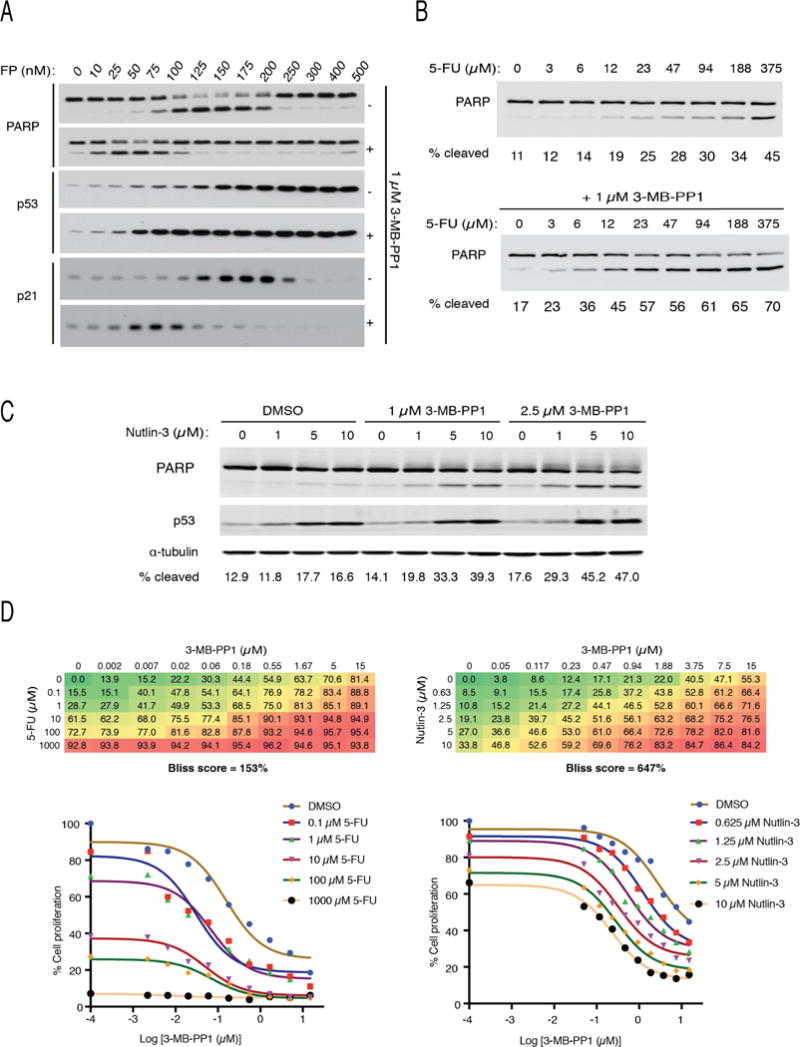
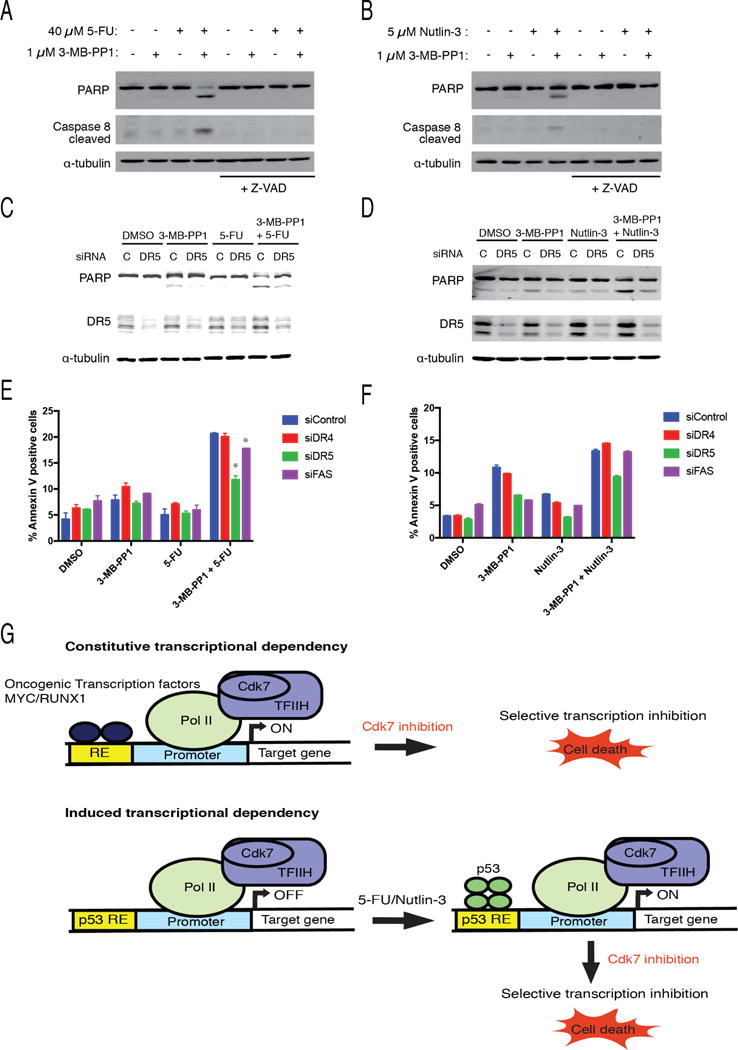
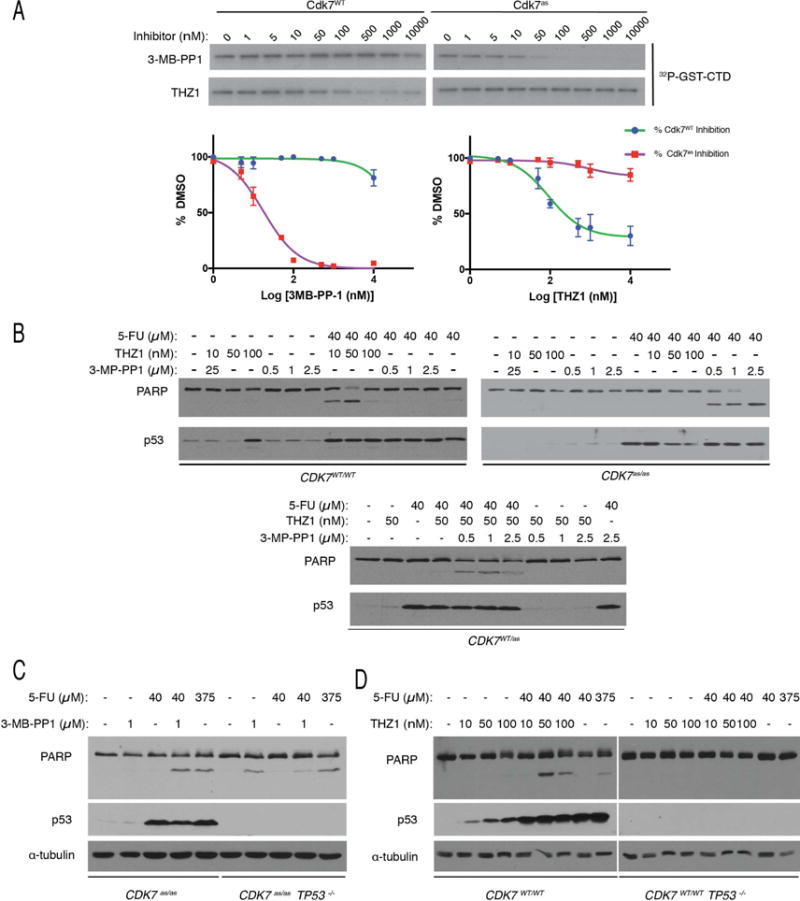
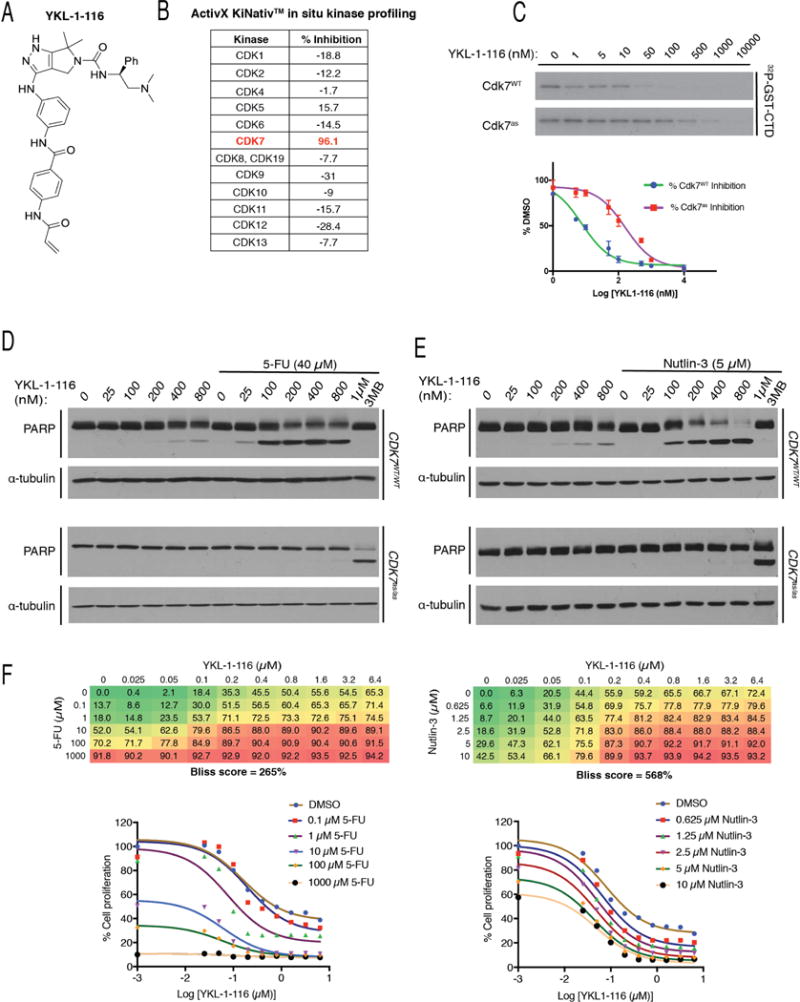
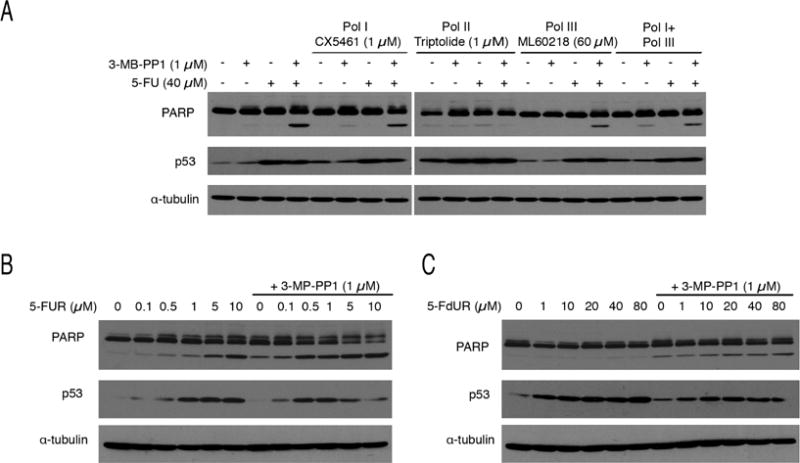
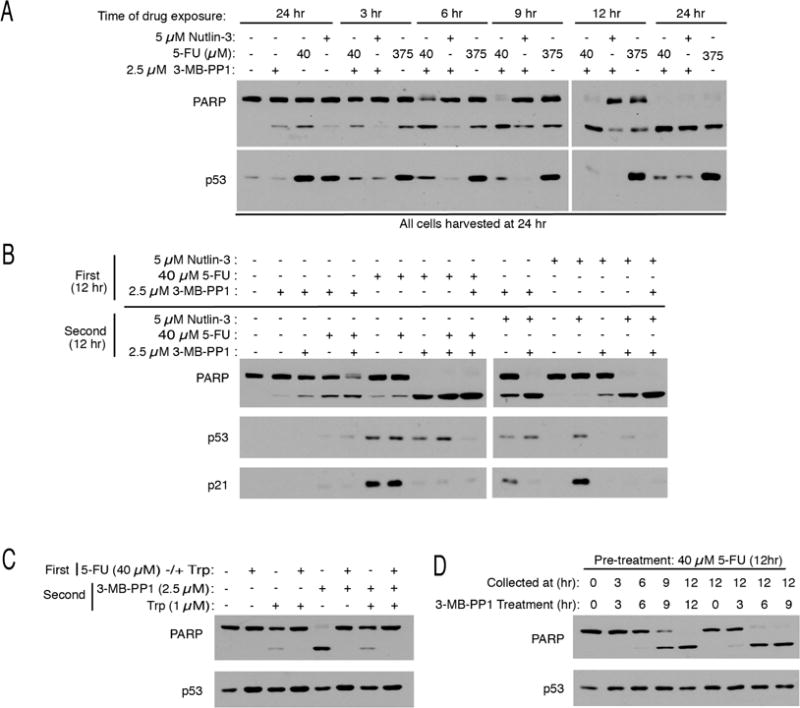
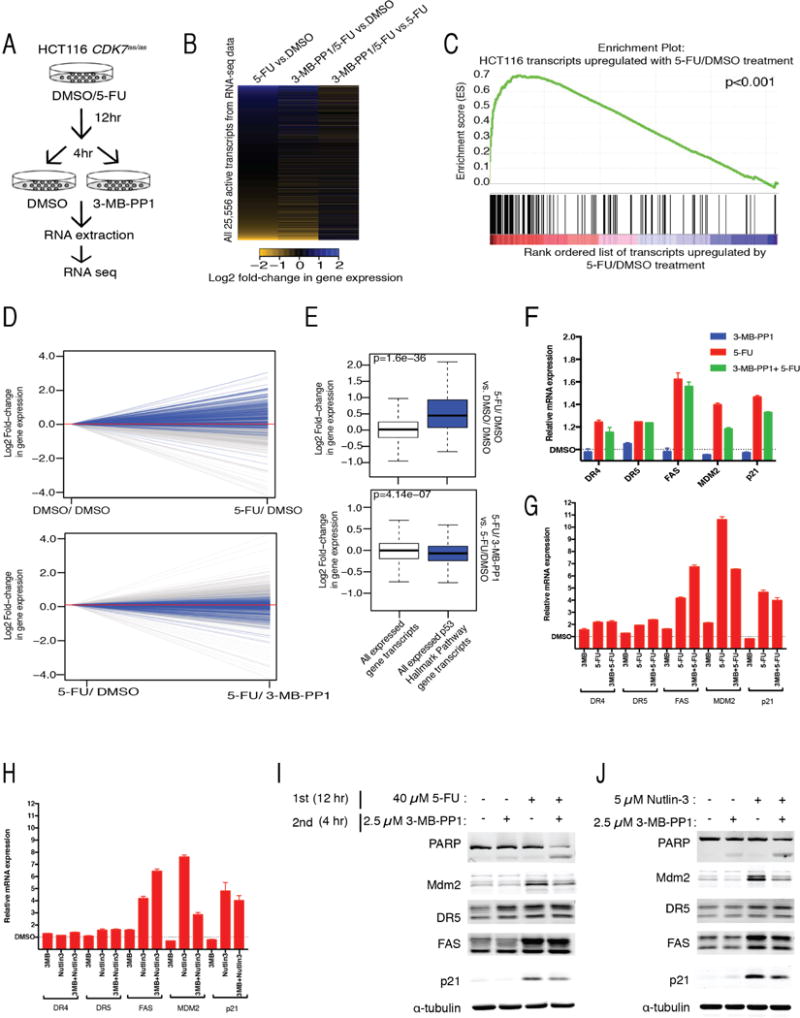
Cdk7, the CDK-activating kinase and transcription factor IIH component, is a target of inhibitors that kill cancer cells by exploiting tumor-specific transcriptional dependencies. However, whereas selective inhibition of analog-sensitive (AS) Cdk7 in colon cancer-derived cells arrests division and disrupts transcription, it does not by itself trigger apoptosis efficiently. Here, we show that p53 activation by 5-fluorouracil or nutlin-3 synergizes with a reversible Cdk7 inhibitor to induce cell death. Synthetic lethality was recapitulated with covalent inhibitors of wild-type Cdk7, THZ1, or the more selective YKL-1-116. The effects were allele specific; a CDK7 mutation conferred both sensitivity to bulky adenine analogs and resistance to covalent inhibitors. Non-transformed colon epithelial cells were resistant to these combinations, as were cancer-derived cells with p53-inactivating mutations. Apoptosis was dependent on death receptor DR5, a p53 transcriptional target whose expression was refractory to Cdk7 inhibition. Therefore, p53 activation induces transcriptional dependency to sensitize cancer cells to Cdk7 inhibition.
Cdk7,即 CDK 激活激酶和转录因子 IIH 成分,是一种抑制剂的靶标,这些抑制剂通过利用肿瘤特异性转录依赖性来杀死癌细胞。然而,尽管在源自结肠癌的细胞中选择性抑制模拟物敏感(AS)Cdk7 会阻止细胞分裂并破坏转录,但它本身并不能有效地触发细胞凋亡。在这里,我们表明,5-氟尿嘧啶或 nutlin-3 激活 p53 与可逆的 Cdk7 抑制剂协同作用以诱导细胞死亡。用野生型 Cdk7 的共价抑制剂 THZ1 或更具选择性的 YKL-1-116 重现了合成致死性。这些效果具有等位基因特异性;CDK7 突变赋予了对大体积腺嘌呤类似物的敏感性和对共价抑制剂的抗性。未转化的结肠上皮细胞对这些组合具有抗性,具有 p53 失活突变的癌症衍生细胞也是如此。细胞凋亡依赖于死亡受体 DR5,DR5 是 p53 的转录靶标,其表达对 Cdk7 抑制具有抗性。因此,p53 激活诱导转录依赖性,使癌细胞对 Cdk7 抑制敏感。