Parihar Neha, Nandi Sisir
Division of Pharmaceutical Chemistry, Global Institute of Pharmaceutical Education and Research, Affiliated to Uttarakhand Technical University, Kashipur, 244713 India.
Springerplus. 2015 Dec 29;4(1):819. doi: 10.1186/s40064-015-1593-3. eCollection 2015.
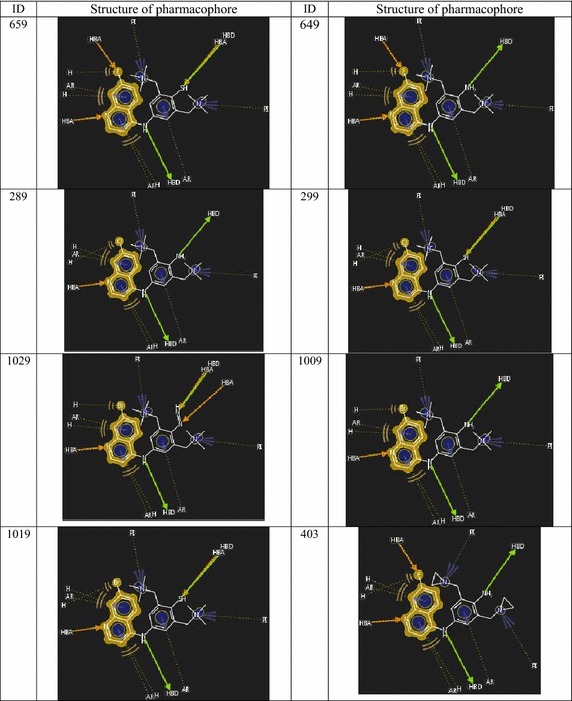
There are very few studies for combinatorial library design and high throughput screening of 4-anilinoquinoline antimalarial compounds having activities against parasitic strain of . Therefore, an attempt has been made in the present paper to design potent lead compounds in this congener utilizing quantitative structure activity relationship utilizing theoretical molecular descriptors. QSAR models for a series of 4-anilinoquinolines considering various theoretical molecular descriptors including topological, constitutional, geometrical, functional group and atom-centered fragments has been carried out by stepwise forward-backward variable selections assimilating multiple linear regression (MLR) methods showing the topological indices contribute maximum impact on parasitic strain. A combinatorial library of 2160 compounds has been generated and finally, 16 compounds were screened through high throughput screening as promising 4-anilinoquinoline antimalarial hits based on their predicted activities utilizing topological descriptor based validated QSAR model. Highly predicted active compounds were then undergone for pharmacophore modeling to predict mode of binding and to optimize leads having greater affinity towards malarial parasitic strain.
针对具有抗疟原虫菌株活性的4-苯胺基喹啉抗疟化合物的组合库设计和高通量筛选的研究非常少。因此,本文尝试利用理论分子描述符,通过定量构效关系来设计该同系物中的有效先导化合物。考虑了包括拓扑、结构、几何、官能团和原子中心片段等各种理论分子描述符的一系列4-苯胺基喹啉的QSAR模型,已通过逐步向前-向后变量选择结合多元线性回归(MLR)方法进行构建,结果表明拓扑指数对疟原虫菌株的影响最大。已生成一个包含2160种化合物的组合库,最后,基于利用基于拓扑描述符的经过验证的QSAR模型预测的活性,通过高通量筛选筛选出16种化合物作为有前景的4-苯胺基喹啉抗疟命中物。然后对预测活性高的化合物进行药效团建模,以预测结合模式并优化对疟原虫菌株具有更高亲和力的先导物。