Zhao Xin, Gao Shouguo, Wu Zhijie, Kajigaya Sachiko, Feng Xingmin, Liu Qingguo, Townsley Danielle M, Cooper James, Chen Jinguo, Keyvanfar Keyvan, Fernandez Ibanez Maria Del Pilar, Wang Xujing, Young Neal S
Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD.
Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, People's Republic of China.
Blood. 2017 Dec 21;130(25):2762-2773. doi: 10.1182/blood-2017-08-803353. Epub 2017 Oct 13.
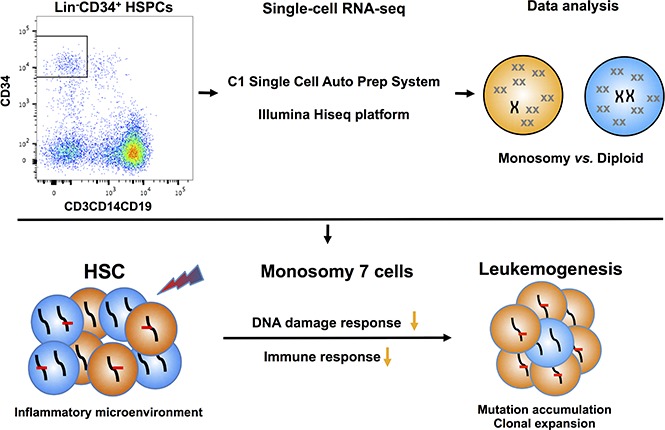
Cancer cells frequently exhibit chromosomal abnormalities. Specific cytogenetic aberrations often are predictors of outcome, especially in hematologic neoplasms, such as monosomy 7 in myeloid malignancies. The functional consequences of aneuploidy at the cellular level are difficult to assess because of a lack of convenient markers to distinguish abnormal from diploid cells. We performed single-cell RNA sequencing (scRNA-seq) to study hematopoietic stem and progenitor cells from the bone marrow of 4 healthy donors and 5 patients with bone marrow failure and chromosome gain or loss. In total, transcriptome sequences were obtained from 391 control cells and 588 cells from patients. We characterized normal hematopoiesis as binary differentiation from stem cells to erythroid and myeloid-lymphoid pathways. Aneuploid cells were distinguished from diploid cells in patient samples by computational analyses of read fractions and gene expression of individual chromosomes. We confirmed assignment of aneuploidy to individual cells quantitatively, by copy-number variation, and qualitatively, by loss of heterozygosity. When we projected patients' single cells onto the map of normal hematopoiesis, diverse patterns were observed, broadly reflecting clinical phenotypes. Patients' monosomy 7 cells showed downregulation of genes involved in immune response and DNA damage checkpoint and apoptosis pathways, which may contribute to the clonal expansion of monosomy 7 cells with accumulated gene mutations. scRNA-seq is a powerful technique through which to infer the functional consequences of chromosome gain and loss and explore gene targets for directed therapy.
癌细胞常常表现出染色体异常。特定的细胞遗传学畸变往往是预后的预测指标,尤其是在血液系统肿瘤中,比如髓系恶性肿瘤中的7号染色体单体。由于缺乏方便的标记来区分异常细胞和二倍体细胞,因此在细胞水平评估非整倍体的功能后果很困难。我们进行了单细胞RNA测序(scRNA-seq),以研究来自4名健康供体和5名骨髓衰竭且有染色体增减的患者骨髓中的造血干细胞和祖细胞。总共从391个对照细胞和患者的588个细胞中获得了转录组序列。我们将正常造血特征化为从干细胞到红系和髓系-淋巴系途径的二元分化。通过对单个染色体的读数分数和基因表达进行计算分析,在患者样本中区分了非整倍体细胞和二倍体细胞。我们通过拷贝数变异在数量上以及通过杂合性缺失在质量上确认了非整倍体在单个细胞中的归属。当我们将患者的单细胞投射到正常造血图谱上时,观察到了不同的模式,大致反映了临床表型。患者的7号染色体单体细胞显示出参与免疫反应、DNA损伤检查点和凋亡途径的基因下调,这可能有助于具有累积基因突变的7号染色体单体细胞的克隆扩增。scRNA-seq是一种强大的技术,通过它可以推断染色体增减的功能后果并探索定向治疗的基因靶点。