Department of Cell Physiology and Molecular Biophysics, Center for Membrane Protein Research, Texas Tech University Health Sciences Center, Lubbock, TX.
School of Biological Sciences, Illinois State University, Normal, IL.
J Gen Physiol. 2017 Nov 6;149(11):1009-1028. doi: 10.1085/jgp.201711827. Epub 2017 Oct 13.
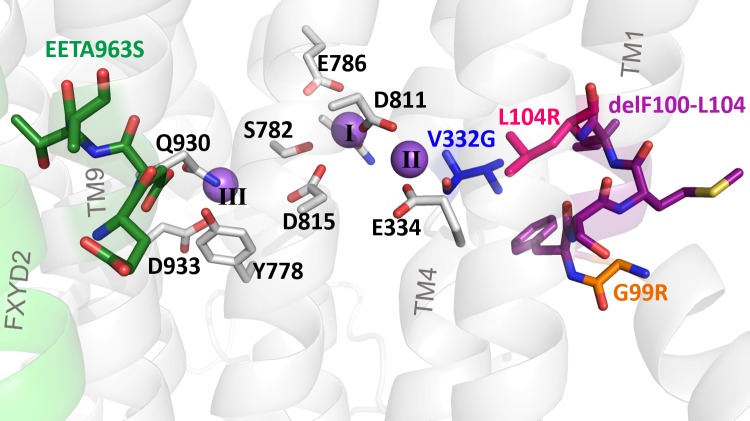
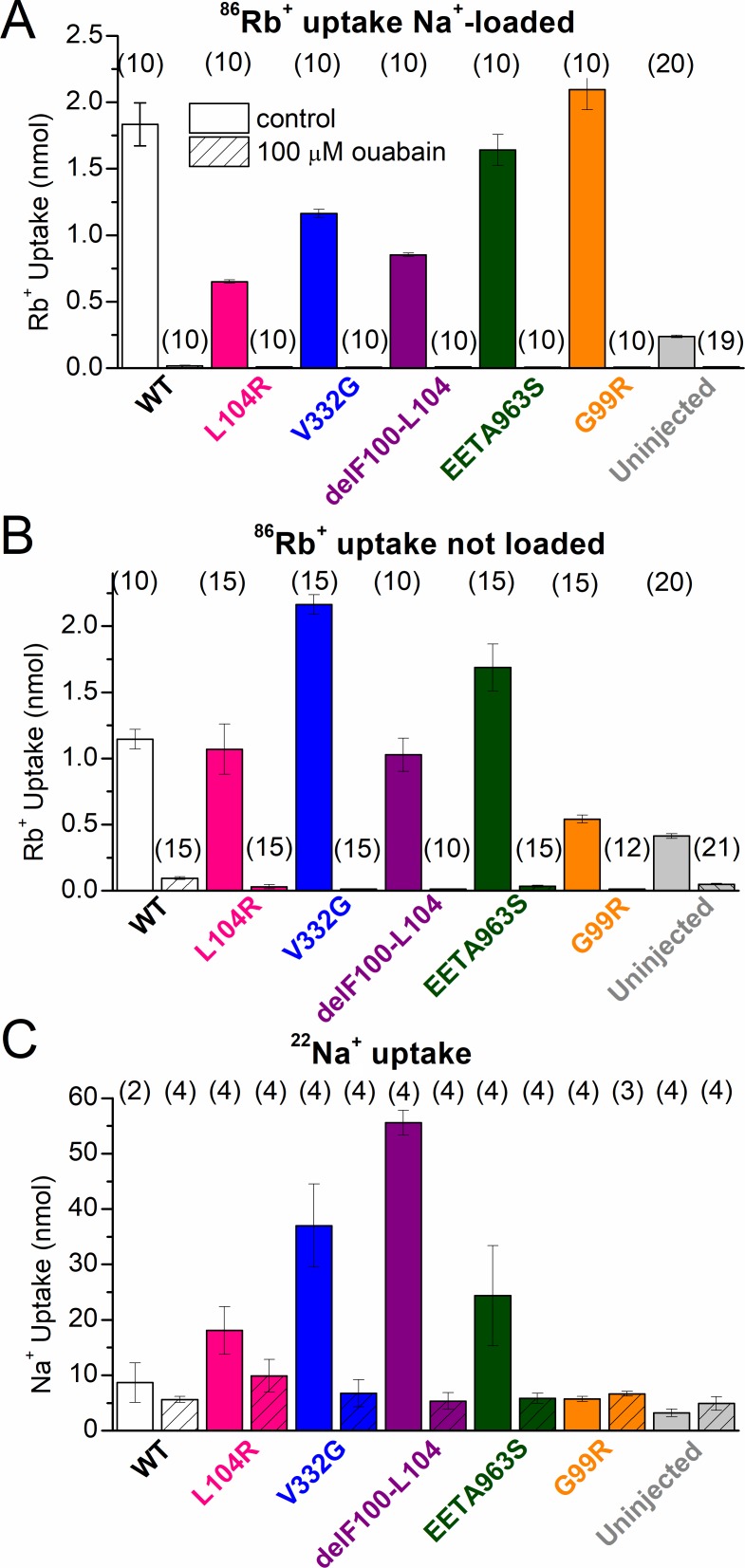
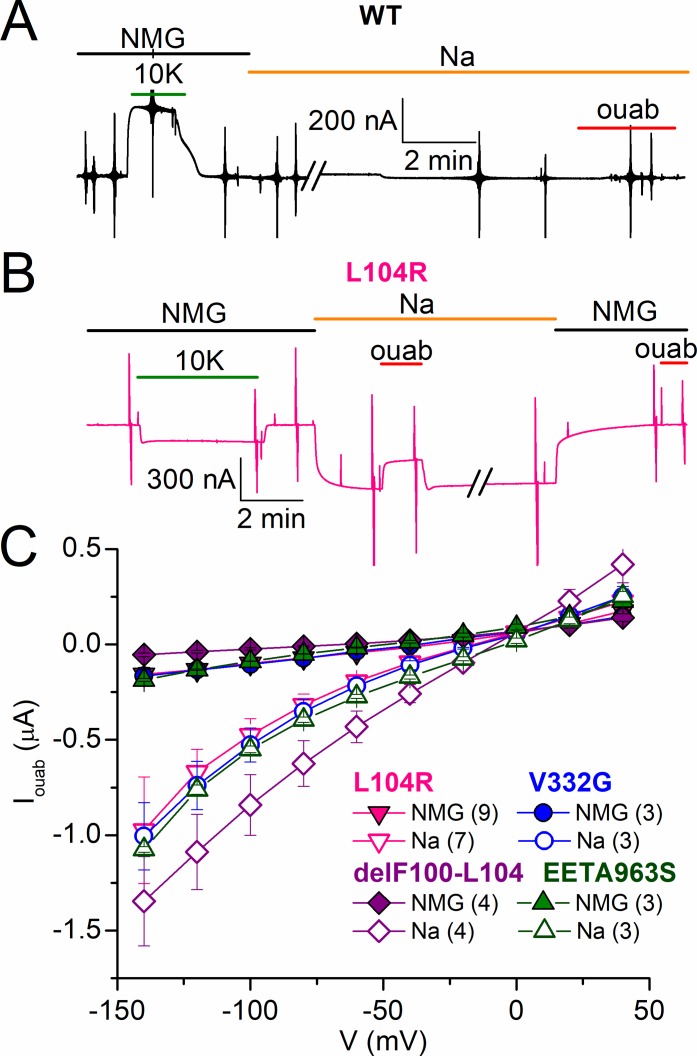
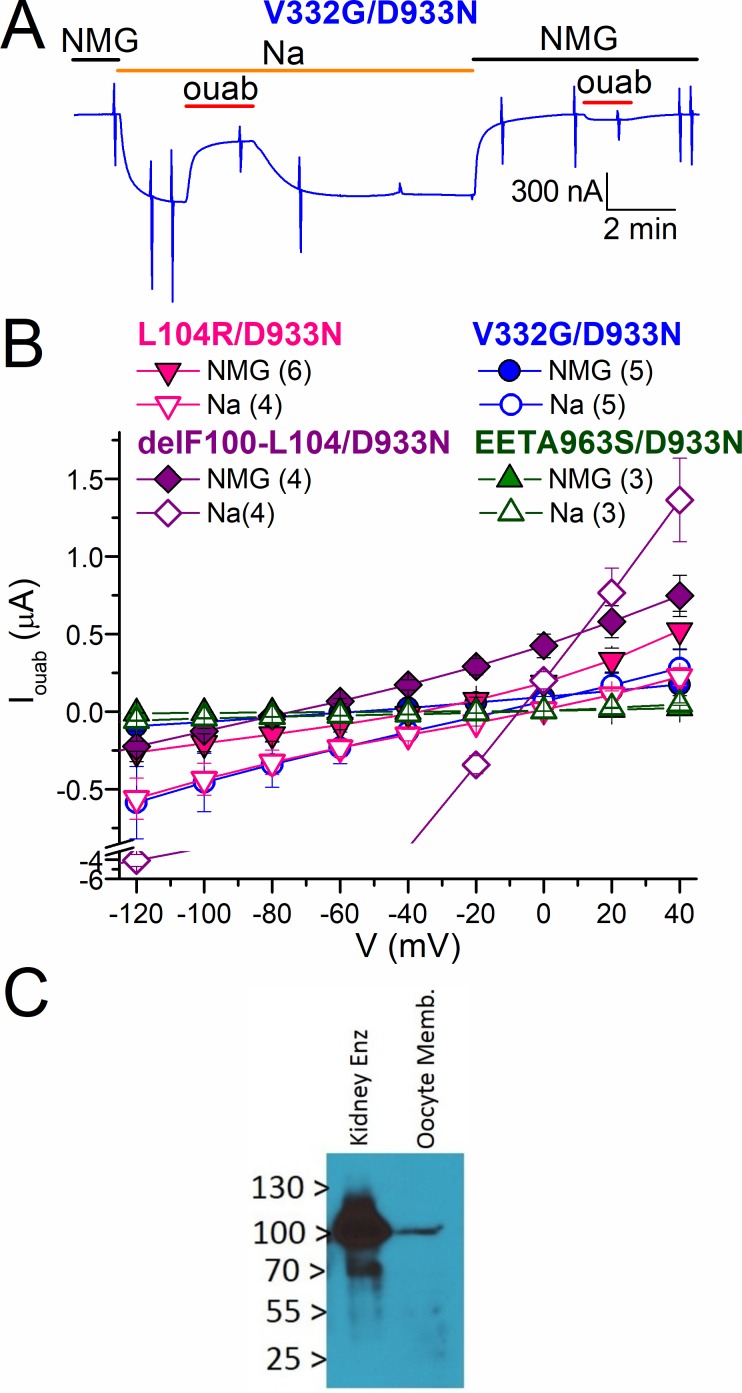
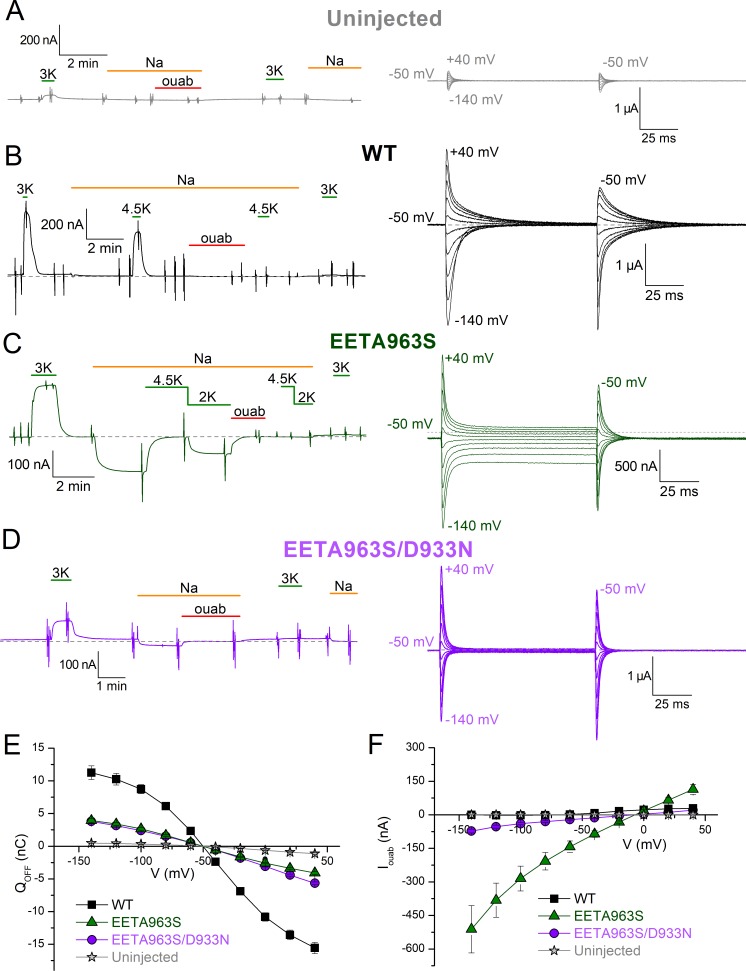
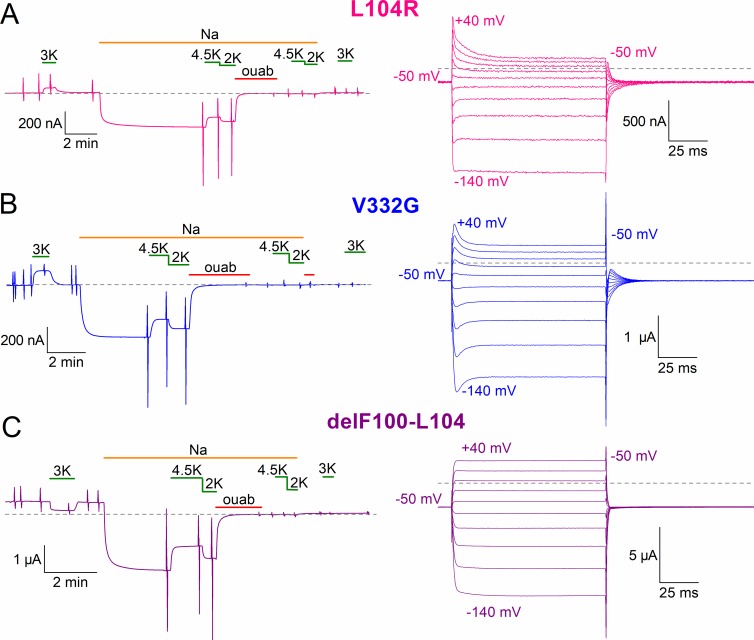
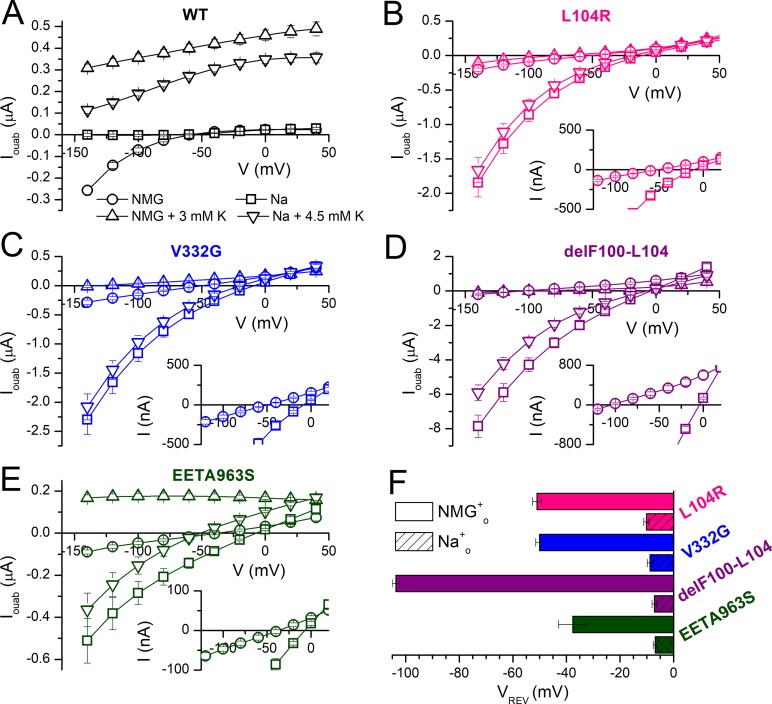
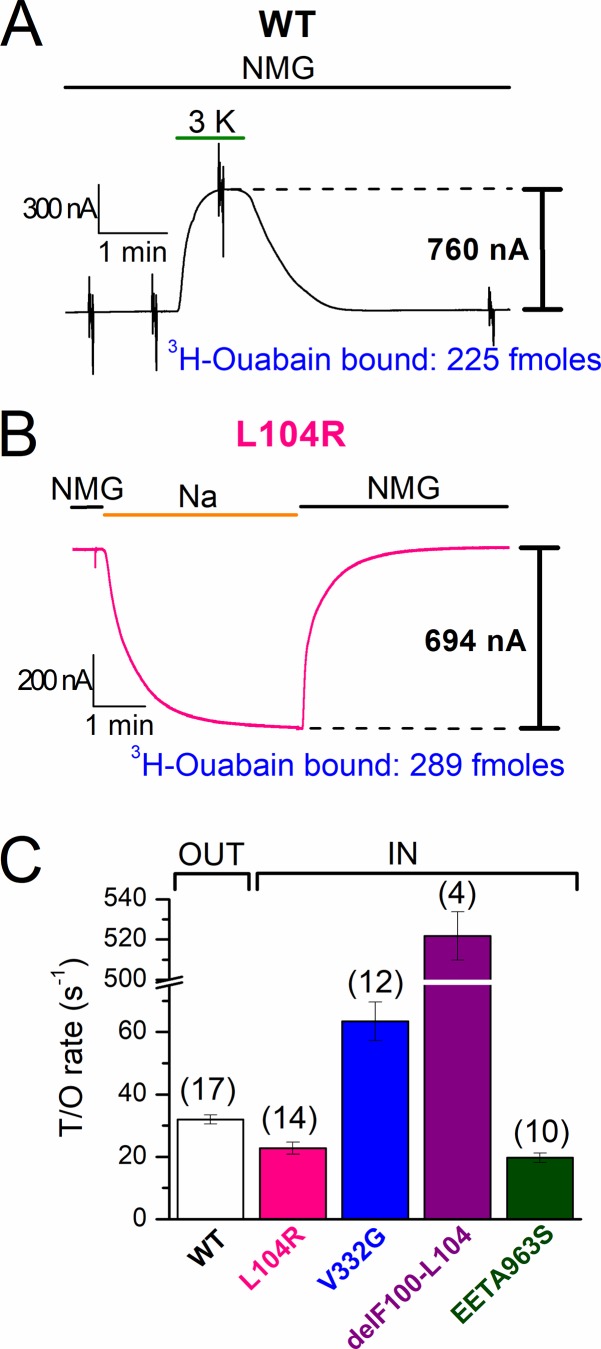
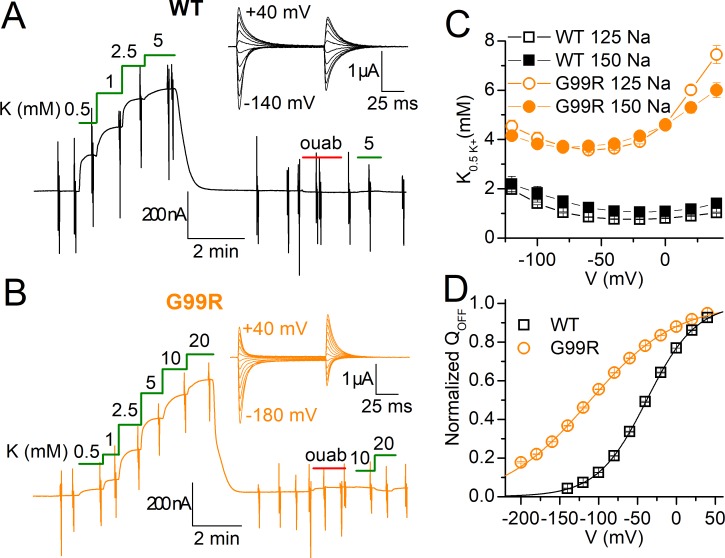
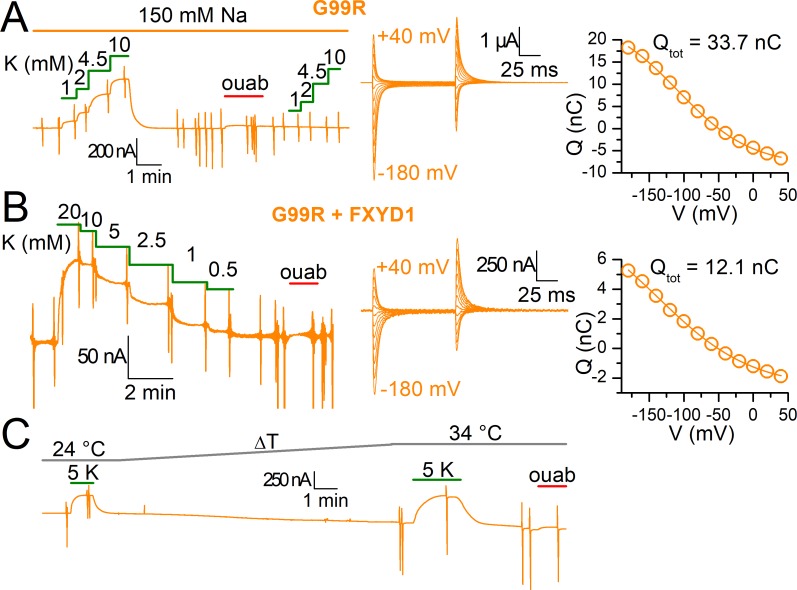
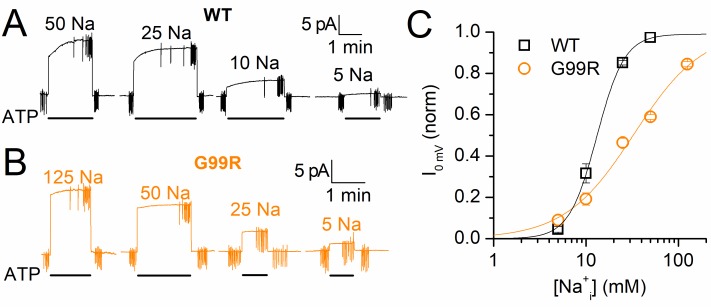
Primary aldosteronism, a condition in which too much aldosterone is produced and that leads to hypertension, is often initiated by an aldosterone-producing adenoma within the zona glomerulosa of the adrenal cortex. Somatic mutations of ATP1A1, encoding the Na/K pump α1 subunit, have been found in these adenomas. It has been proposed that a passive inward current transported by several of these mutant pumps is a "gain-of-function" activity that produces membrane depolarization and concomitant increases in aldosterone production. Here, we investigate whether the inward current through mutant Na/K pumps is large enough to induce depolarization of the cells that harbor them. We first investigate inward currents induced by these mutations in Na/K pumps expressed in oocytes and find that these inward currents are similar in amplitude to wild-type outward Na/K pump currents. Subsequently, we perform a detailed functional evaluation of the human Na/K pump mutants L104R, delF100-L104, V332G, and EETA963S expressed in oocytes. By combining two-electrode voltage clamp with [H]ouabain binding, we measure the turnover rate of these inward currents and compare it to the turnover rate for outward current through wild-type pumps. We find that the turnover rate of the inward current through two of these mutants (EETA963S and L104R) is too small to induce significant cell depolarization. Electrophysiological characterization of another hyperaldosteronism-inducing mutation, G99R, reveals the absence of inward currents under many different conditions, including in the presence of the regulator FXYD1 as well as with mammalian ionic concentrations and body temperatures. Instead, we observe robust outward currents, but with significantly reduced affinities for intracellular Na and extracellular K Collectively, our results point to loss-of-function as the common mechanism for the hyperaldosteronism induced by these Na/K pump mutants.
原发性醛固酮增多症是一种由于醛固酮分泌过多而导致高血压的疾病,其通常由肾上腺皮质球状带中的醛固酮产生腺瘤引起。在这些腺瘤中已经发现了编码钠钾泵α1亚基的 ATP1A1 的体细胞突变。有人提出,这些突变泵所携带的内向离子流是一种“功能获得”活性,可导致膜去极化并伴随醛固酮产生增加。在这里,我们研究了通过突变钠钾泵的内向电流是否足以诱导携带它们的细胞去极化。我们首先在卵母细胞中表达的这些突变钠钾泵中研究了这些突变引起的内向电流,发现这些内向电流的幅度与野生型外向钠钾泵电流相似。随后,我们在卵母细胞中对人钠钾泵突变体 L104R、delF100-L104、V332G 和 EETA963S 进行了详细的功能评估。通过将双电极电压钳与[H]哇巴因结合,我们测量了这些内向电流的周转率,并将其与外向电流通过野生型泵的周转率进行了比较。我们发现,其中两种突变体(EETA963S 和 L104R)的内向电流周转率太小,无法引起明显的细胞去极化。另一种引起高醛固酮血症的突变 G99R 的电生理特性研究表明,在许多不同的条件下,包括存在调节剂 FXYD1 以及哺乳动物离子浓度和体温下,都没有内向电流。相反,我们观察到强大的外向电流,但对细胞内 Na 和细胞外 K 的亲和力明显降低。总的来说,我们的结果表明,这些钠钾泵突变体引起的高醛固酮血症的共同机制是功能丧失。