Aschard Hugues, Guillemot Vincent, Vilhjalmsson Bjarni, Patel Chirag J, Skurnik David, Ye Chun J, Wolpin Brian, Kraft Peter, Zaitlen Noah
Centre de Bioinformatique, Biostatistique et Biologie Intégrative (C3BI), Institut Pasteur, Paris, France.
Department of Epidemiology, Harvard TH Chan School of Public Health, Boston, Massachusetts, USA.
Nat Genet. 2017 Dec;49(12):1789-1795. doi: 10.1038/ng.3975. Epub 2017 Oct 16.
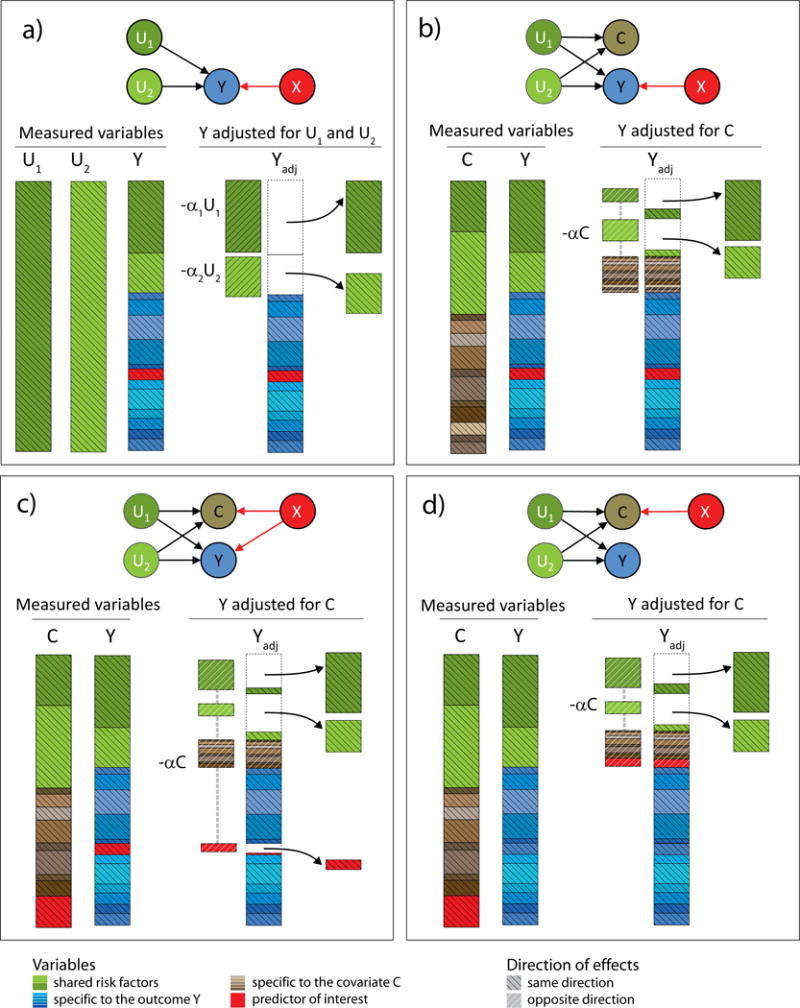
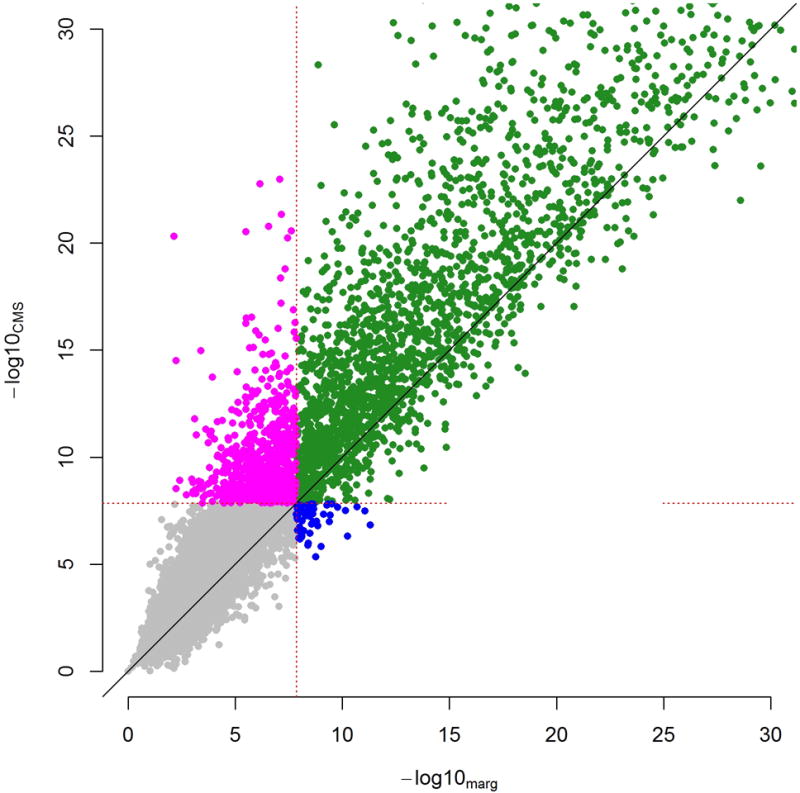
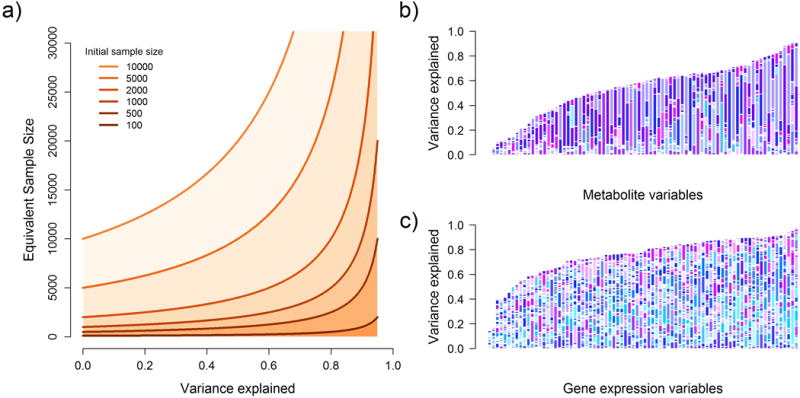
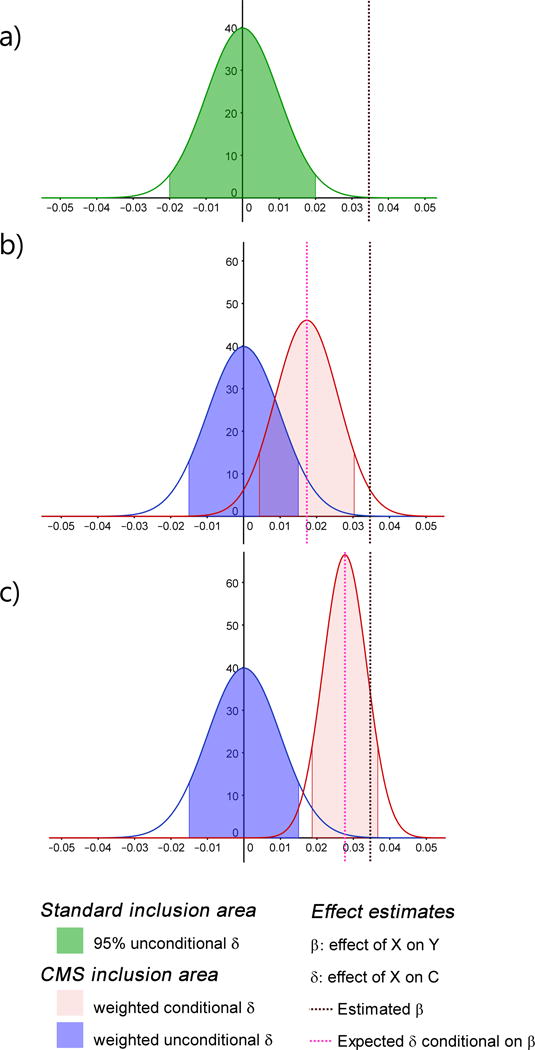
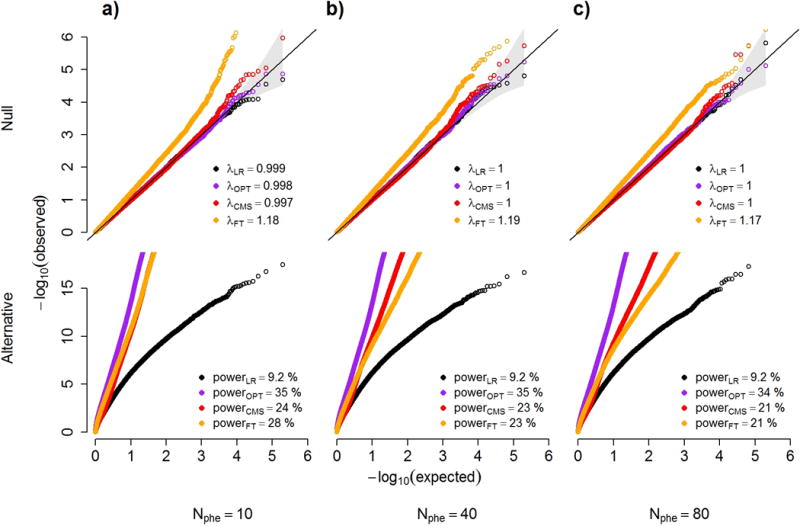
Testing for associations in big data faces the problem of multiple comparisons, wherein true signals are difficult to detect on the background of all associations queried. This difficulty is particularly salient in human genetic association studies, in which phenotypic variation is often driven by numerous variants of small effect. The current strategy to improve power to identify these weak associations consists of applying standard marginal statistical approaches and increasing study sample sizes. Although successful, this approach does not leverage the environmental and genetic factors shared among the multiple phenotypes collected in contemporary cohorts. Here we developed covariates for multiphenotype studies (CMS), an approach that improves power when correlated phenotypes are measured on the same samples. Our analyses of real and simulated data provide direct evidence that correlated phenotypes can be used to achieve increases in power to levels often surpassing the power gained by a twofold increase in sample size.
在大数据中检测关联面临多重比较问题,即在所有查询的关联背景下,真实信号难以检测。这种困难在人类基因关联研究中尤为突出,其中表型变异通常由众多小效应变体驱动。目前提高识别这些弱关联能力的策略包括应用标准的边际统计方法和增加研究样本量。尽管这种方法取得了成功,但它没有利用当代队列中收集的多个表型之间共享的环境和遗传因素。在此,我们开发了多表型研究协变量(CMS),这是一种在对相同样本测量相关表型时提高能力的方法。我们对真实数据和模拟数据的分析提供了直接证据,表明相关表型可用于将能力提高到通常超过样本量翻倍所获得的能力水平。