Division of Animal Sciences, University of Missouri, Columbia, MO, 65211, USA.
Informatics Institute, University of Missouri, Columbia, MO, 65211, USA.
BMC Genomics. 2017 Oct 18;18(1):799. doi: 10.1186/s12864-017-4196-2.
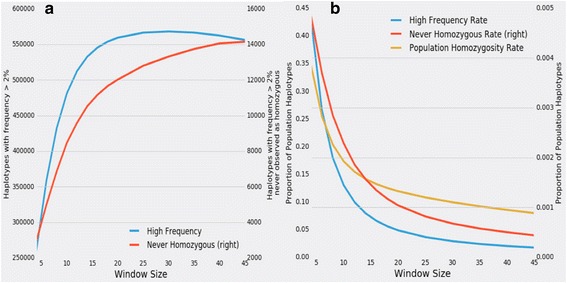
If unmanaged, high rates of inbreeding in livestock populations adversely impact their reproductive fitness. In beef cattle, historical selection strategies have increased the frequency of several segregating fatal autosomal recessive polymorphisms. Selective breeding has also decreased the extent of haplotypic diversity genome-wide. By identifying haplotypes for which homozygotes are not observed but would be expected based on their frequency, candidates for developmentally lethal recessive loci can be localized. This analysis comes without the need for observation of the loss-associated phenotype (e.g., failure to implant, first trimester abortion, deformity at birth). In this study, haplotypes were estimated for 3961 registered Angus individuals using 52,545 SNP loci using findhap v2, which exploited the complex pedigree among the individuals in this population.
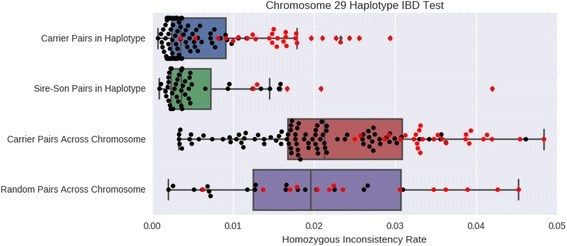
Seven loci were detected to possess haplotypes that were not observed in homozygous form despite a sufficiently high frequency and pedigree-based expectation of homozygote occurrence. These haplotypes were identified as candidates for harboring autosomal recessive lethal alleles. Of the genotyped individuals, 109 were resequenced to an average 27X depth of coverage to identify putative loss-of-function alleles genome-wide and had variants called using a custom in-house developed pipeline. For the candidate lethal-harboring haplotypes present in these bulls, sequence-called genotypes were used to identify concordant variants. In addition, whole-genome sequence imputation of variants was performed into the set of 3961 genotyped animals using the 109 resequenced animals to identify candidate lethal recessive variants at the seven loci. Following the imputation, no variants were identified that were fully concordant with the marker-based diplotypes.
Selective breeding programs could utilize the predicted lethal haplotypes associated with SNP genotypes. Sequencing and other methods for identifying the causal variants underlying these haplotypes can allow for more efficient methods of management such as gene editing. These two methods in total will reduce the negative impacts of inbreeding on fertility and maximize overall genetic gains.
如果牲畜种群中的近亲繁殖得不到有效控制,会对其繁殖适应性产生不利影响。在肉牛中,历史上的选择策略增加了几个分离的致命常染色体隐性多态性的频率。选择性繁殖也降低了全基因组范围内单倍型多样性的程度。通过识别那些没有观察到纯合子但根据其频率预期会存在的单倍型,可以定位到发育致死的隐性基因座候选者。这种分析不需要观察到与丢失相关的表型(例如,植入失败、早期流产、出生时畸形)。在这项研究中,使用 findhap v2 对 3961 头注册的安格斯个体的 52545 个 SNP 位点进行了单倍型估计,该软件利用了该群体中个体之间复杂的系谱。
尽管存在足够高的频率和基于系谱的纯合子发生预期,但有 7 个基因座被检测到具有未观察到的纯合单倍型。这些单倍型被确定为携带常染色体隐性致死等位基因的候选者。在这些个体中,有 109 个个体进行了重新测序,平均测序深度为 27X,以鉴定全基因组范围内的潜在功能丧失等位基因,并使用内部开发的定制管道进行了变体调用。对于这些公牛中存在的候选致死性单倍型,使用序列调用基因型来识别一致的变体。此外,使用这 109 个重测序动物对 3961 个已分型动物进行了全基因组序列的变异体导入,以鉴定这 7 个基因座上的候选致死性隐性变异体。在导入后,没有发现与基于标记的二倍型完全一致的变异体。
选择性繁殖计划可以利用与 SNP 基因型相关的预测致死性单倍型。用于识别这些单倍型下因果变异的测序和其他方法可以为管理提供更有效的方法,例如基因编辑。这两种方法总共可以减少近亲繁殖对生育能力的负面影响,并最大限度地提高整体遗传增益。