Chaput Ludovic, Mouawad Liliane
Chemistry, Modelling and Imaging for Biology (CMIB), Institut Curie - PSL Research University, Bât 112, Centre Universitaire, 91405, Orsay Cedex, France.
Paris-Sud University, Orsay, France.
J Cheminform. 2017 Jun 12;9(1):37. doi: 10.1186/s13321-017-0227-x.
In drug design, an efficient structure-based optimization of a ligand needs the precise knowledge of the protein-ligand interactions. In the absence of experimental information, docking programs are necessary for ligand positioning, and the choice of a reliable program is essential for the success of such an optimization. The performances of four popular docking programs, Gold, Glide, Surflex and FlexX, were investigated using 100 crystal structures of complexes taken from the Directory of Useful Decoys-Enhanced database.
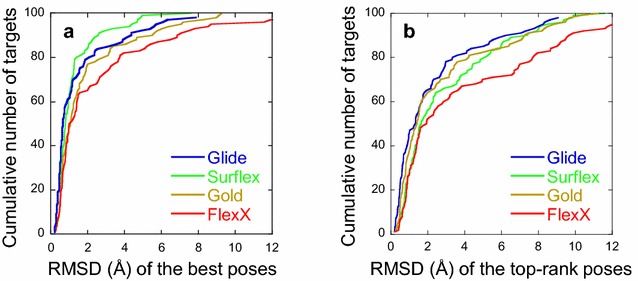
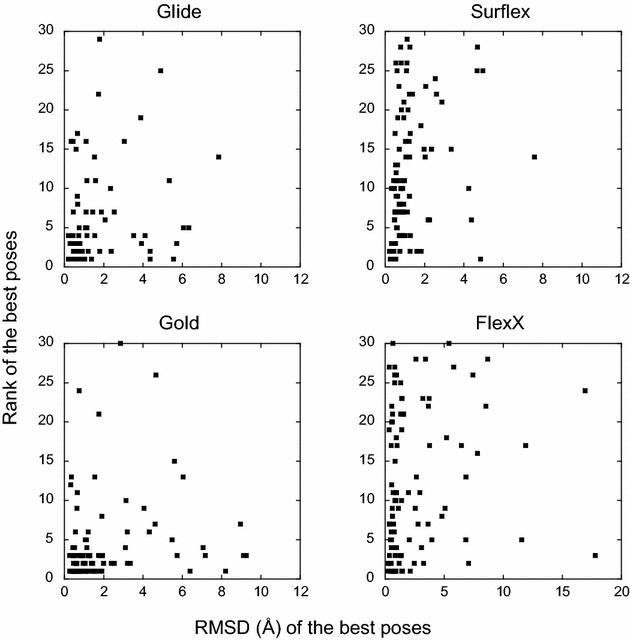
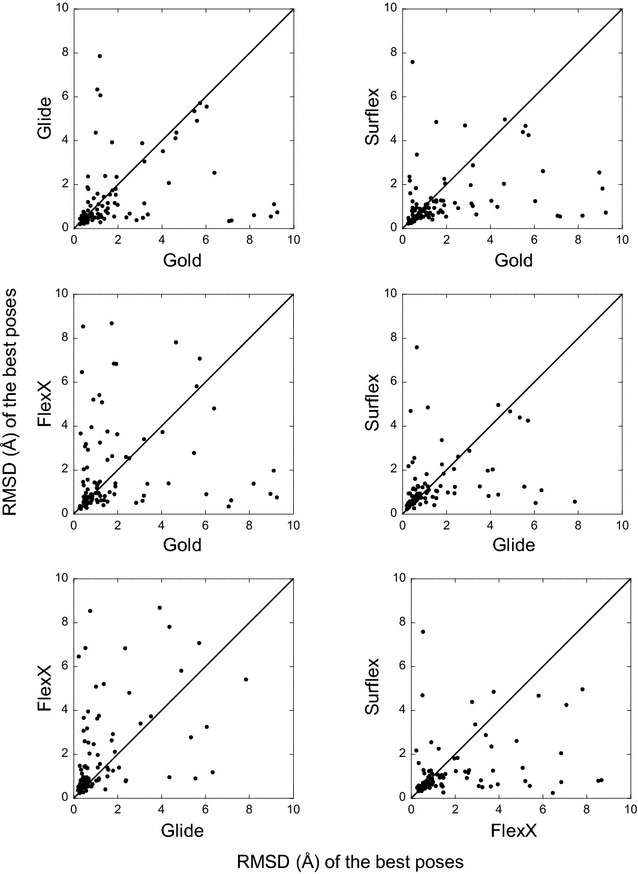
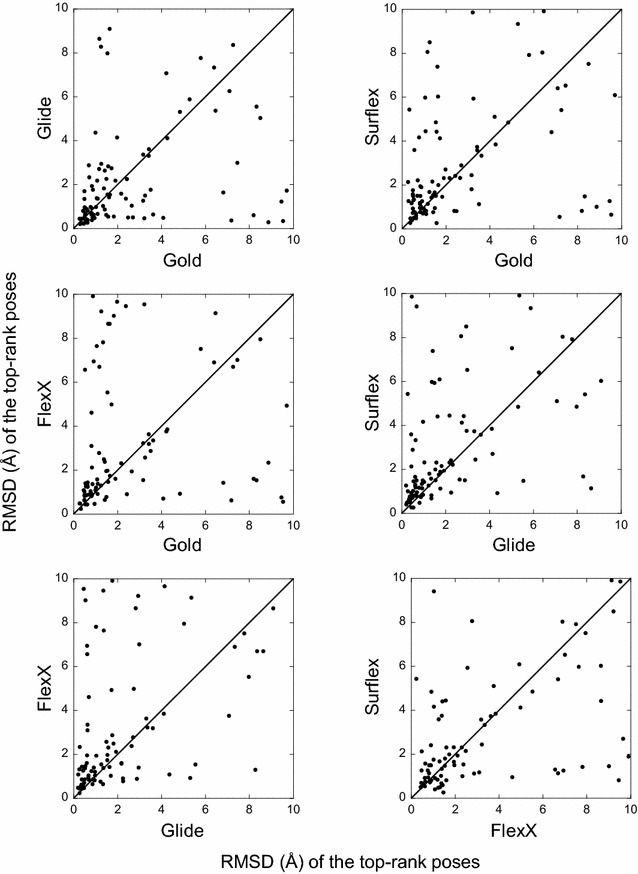
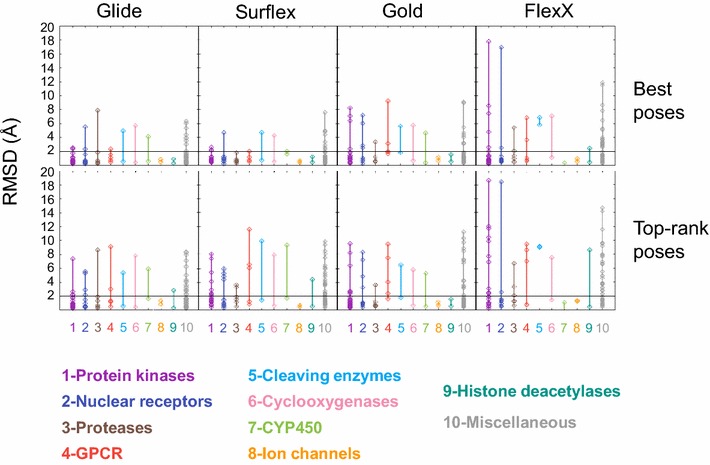
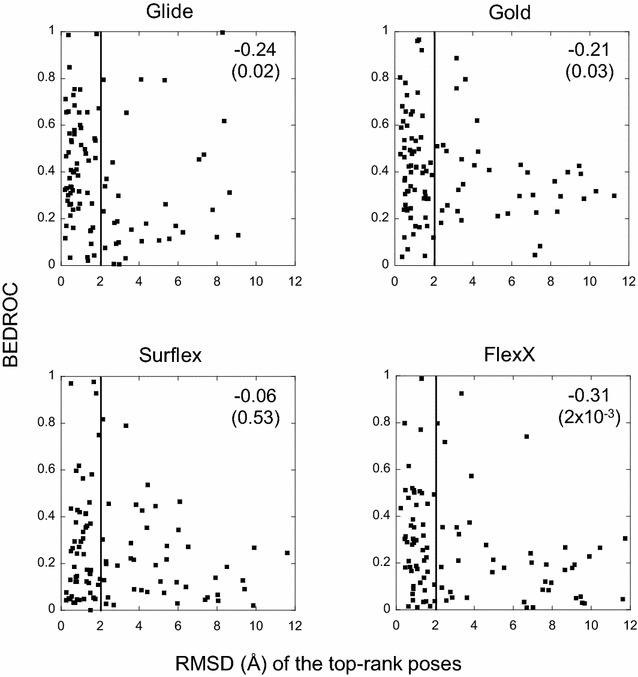
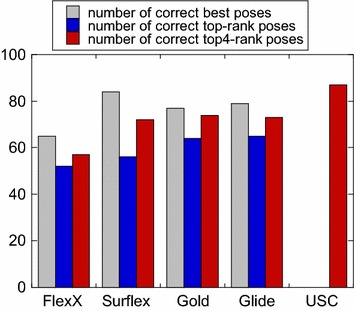
The ligand conformational sampling was rather efficient, with a correct pose found for a maximum of 84 complexes, obtained by Surflex. However, the ranking of the correct poses was not as efficient, with a maximum of 68 top-rank or 75 top-4 rank correct poses given by Glidescore. No relationship was found between either the sampling or the scoring performance of the four programs and the properties of either the targets or the small molecules, except for the number of ligand rotatable bonds. As well, no exploitable relationship was found between each program performance in docking and in virtual screening; a wrong top-rank pose may obtain a good score that allows it to be ranked among the most active compounds and vice versa. Also, to improve the results of docking, the strengths of the programs were combined either by using a rescoring procedure or the United Subset Consensus (USC). Oddly, positioning with Surflex and rescoring with Glidescore did not improve the results. However, USC based on docking allowed us to obtain a correct pose in the top-4 rank for 87 complexes. Finally, nine complexes were scrutinized, because a correct pose was found by at least one program but poorly ranked by all four programs. Contrarily to what was expected, except for one case, this was not due to weaknesses of the scoring functions.
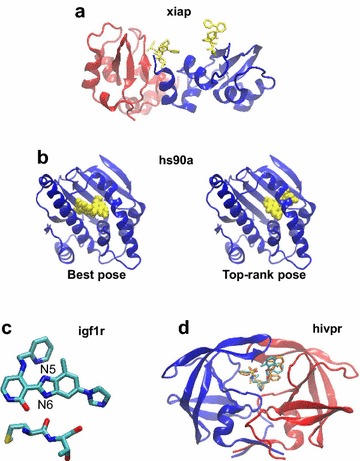
We conclude that the scoring functions should be improved to detect the correct poses, but sometimes their failure may be due to other varied considerations. To increase the chances of success, we recommend to use several programs and combine their results. Graphical abstract Summary of the results obtained by semi-rigid docking of crystallographic ligands. The docking was done on 100 protein-ligand X-ray structures, taken from the DUD-E database, and using four programs, Glide, Gold, Surflex and FlexX. Based on the docking results, we applied our United Subset Consensus method (USC), for which only the top4-rank poses are relevant. The number of complexes for which the best pose is correct, is represented by the gray boxes, the blue and red boxes correspond to the number of complexes with a correct pose ranked as the top 1 or within the top 4. A pose is considered correct when its root-mean-square deviation from the crystal structure is less than 2 Å.
在药物设计中,基于结构对配体进行有效的优化需要精确了解蛋白质 - 配体相互作用。在缺乏实验信息的情况下,对接程序对于配体定位是必要的,而选择一个可靠的程序对于这种优化的成功至关重要。使用从有用诱饵增强数据库目录中获取的100个复合物晶体结构,研究了四种流行对接程序Gold、Glide、Surflex和FlexX的性能。
配体构象采样相当有效,Surflex程序最多为84个复合物找到了正确的构象。然而,正确构象的排名效率不高,Glidescore程序给出的最高排名正确构象为68个,前四名正确构象最多为75个。除了配体可旋转键的数量外,未发现这四个程序的采样或评分性能与靶标或小分子的性质之间存在任何关系。同样,在对接和虚拟筛选中,每个程序的性能之间也未发现可利用的关系;错误的最高排名构象可能获得高分从而使其在最具活性的化合物中排名靠前,反之亦然。此外,为了提高对接结果,通过重新评分程序或联合子集共识(USC)方法来合并程序的优势。奇怪的是,使用Surflex进行定位并使用Glidescore进行重新评分并没有改善结果。然而,基于对接的USC方法使我们能够为87个复合物在前四名中获得正确的构象。最后,对九个复合物进行了仔细研究,因为至少有一个程序找到了正确的构象,但在所有四个程序中排名都很差。与预期相反,除了一个案例外,这并非由于评分函数的缺陷。
我们得出结论,应改进评分函数以检测正确的构象,但有时其失败可能是由于其他各种因素。为了增加成功的机会,我们建议使用多个程序并合并它们的结果。图形摘要:晶体学配体半刚性对接获得的结果总结。对接是在从DUD - E数据库中获取的100个蛋白质 - 配体X射线结构上进行的,并使用了四个程序Glide、Gold、Surflex和FlexX。基于对接结果,我们应用了联合子集共识方法(USC),对于该方法只有前四名的构象是相关的。最佳构象正确的复合物数量由灰色框表示,蓝色和红色框分别对应于正确构象排名为第一或在前四名中的复合物数量。当构象与晶体结构的均方根偏差小于2 Å时,该构象被认为是正确的。