Department of Pharmaceutical Sciences & Technology, Birla Institute of Technology, Mesra, Ranchi, Jharkhand, 835 215, India.
Department of Pharmacy, "Drug Discovery" Laboratory, University of Napoli "Federico II", Via D. Montesano, 49, 80131, Napoli, Italy.
Sci Rep. 2017 Oct 31;7(1):14453. doi: 10.1038/s41598-017-14776-0.
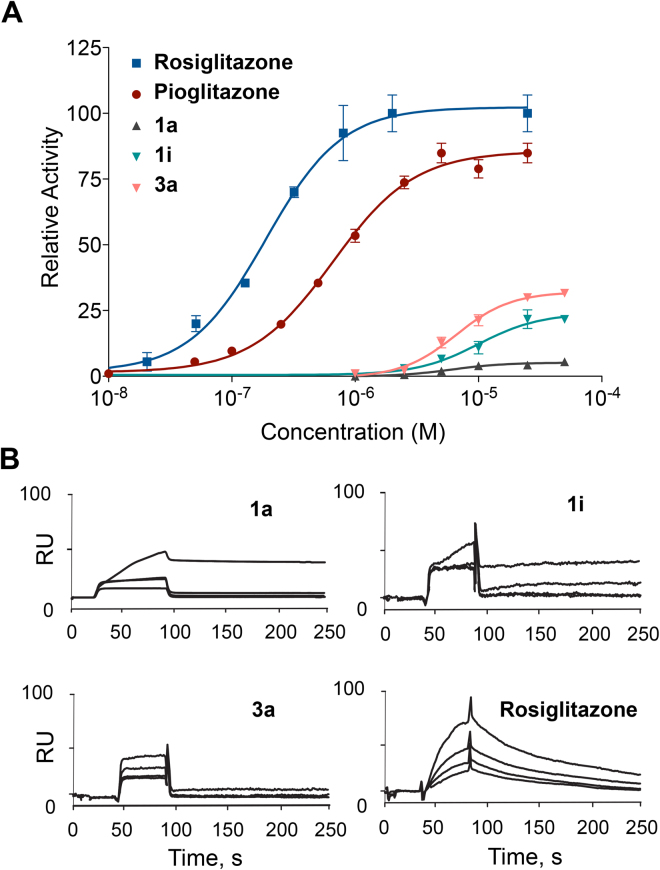
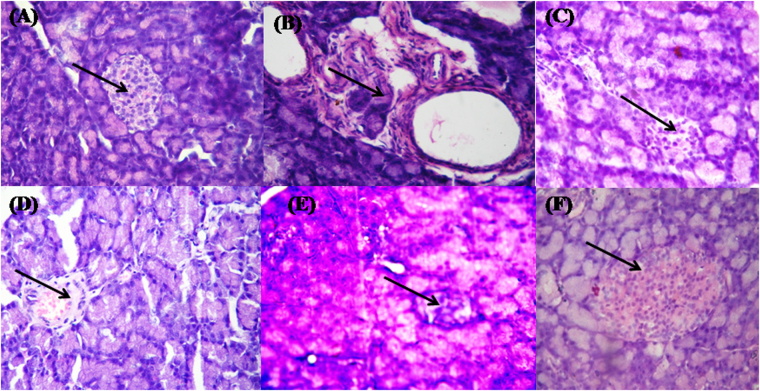
Peroxisome proliferator-activated receptor γ (PPARγ) has received significant attention as a key regulator of glucose and lipid homeostasis. In this study, we synthesized and tested a library of novel 5-benzylidene-thiazolidin-2,4-dione (BTZD) derivatives bearing a substituent on nitrogen of TZD nucleus (compounds 1a-1k, 2i-10i, 3a, 6a, and 8a-10a). Three compounds (1a, 1i, and 3a) exhibited selectivity towards PPARγ and were found to be weak to moderate partial agonists. Surface Plasmon Resonance (SPR) results demonstrated binding affinity of 1a, 1i and 3a towards PPARγ. Furthermore, docking experiments revealed that BTZDs interact with PPARγ through a distinct binding mode, forming primarily hydrophobic contacts with the ligand-binding pocket (LBD) without direct H-bonding interactions to key residues in H12 that are characteristic of full agonists. In addition, 1a, 1i and 3a significantly improved hyperglycemia and hyperlipidaemia in streptozotocin-nicotinamide (STZ-NA)-induced diabetic rats at a dose of 36 mg/kg/day administered orally for 15 days. Histopathological investigations revealed that microscopic architecture of pancreatic and hepatic cells improved in BTZDs-treated diabetic rats. These findings suggested that 1a, 1i and 3a are very promising pharmacological agents by selectively targeting PPARγ for further development in the clinical treatment of type 2 diabetes mellitus.
过氧化物酶体增殖物激活受体 γ(PPARγ)作为葡萄糖和脂质稳态的关键调节剂受到了广泛关注。在这项研究中,我们合成并测试了一系列新型 5-亚苄基噻唑烷-2,4-二酮(BTZD)衍生物,这些衍生物在 TZD 核的氮上带有取代基(化合物 1a-1k、2i-10i、3a、6a 和 8a-10a)。有三种化合物(1a、1i 和 3a)对 PPARγ 具有选择性,并且被发现是弱到中等的部分激动剂。表面等离子体共振(SPR)结果表明 1a、1i 和 3a 对 PPARγ 的结合亲和力。此外,对接实验表明 BTZDs 通过独特的结合模式与 PPARγ 相互作用,主要与配体结合口袋(LBD)形成疏水性相互作用,而不与特征为完全激动剂的 H12 中的关键残基形成直接氢键相互作用。此外,在 STZ-NA 诱导的糖尿病大鼠中,1a、1i 和 3a 以 36mg/kg/天的剂量口服给药 15 天,可显著改善高血糖和高血脂。组织病理学研究表明,BTZDs 治疗的糖尿病大鼠的胰腺和肝细胞微观结构得到改善。这些发现表明,1a、1i 和 3a 是非常有前途的药理学药物,通过选择性靶向 PPARγ,为 2 型糖尿病的临床治疗进一步发展提供了可能。