Siper Paige M, De Rubeis Silvia, Trelles Maria Del Pilar, Durkin Allison, Di Marino Daniele, Muratet François, Frank Yitzchak, Lozano Reymundo, Eichler Evan E, Kelly Morgan, Beighley Jennifer, Gerdts Jennifer, Wallace Arianne S, Mefford Heather C, Bernier Raphael A, Kolevzon Alexander, Buxbaum Joseph D
Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY USA.
Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, USA.
Mol Autism. 2017 Oct 24;8:57. doi: 10.1186/s13229-017-0172-6. eCollection 2017.
Haploinsufficiency of the forkhead-box protein P1 () gene leads to a neurodevelopmental disorder termed FOXP1 syndrome. Previous studies in individuals carrying mutations and deletions have described the presence of autism spectrum disorder (ASD) traits, intellectual disability, language impairment, and psychiatric features. The goal of the present study was to comprehensively characterize the genetic and clinical spectrum of FOXP1 syndrome. This is the first study to prospectively examine the genotype-phenotype relationship in multiple individuals with FOXP1 syndrome, using a battery of standardized clinical assessments.
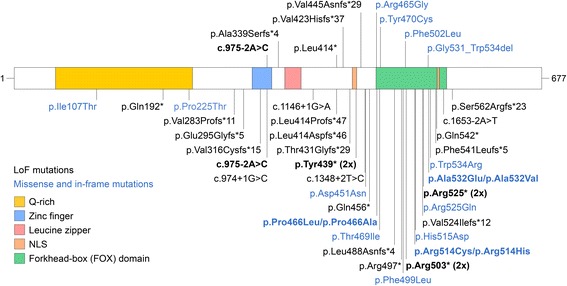
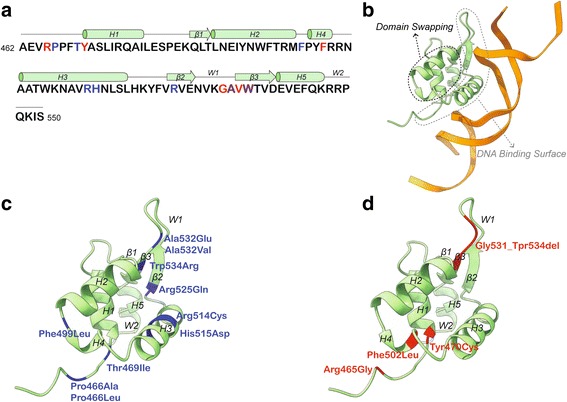
Genetic and clinical data was obtained and analyzed from nine children and adolescents between the ages of 5-17 with mutations in . Phenotypic characterization included gold standard ASD testing and norm-referenced measures of cognition, adaptive behavior, language, motor, and visual-motor integration skills. In addition, psychiatric, medical, neurological, and dysmorphology examinations were completed by a multidisciplinary team of clinicians. A comprehensive review of reported cases was also performed. All missense and in-frame mutations were mapped onto the three-dimensional structure of DNA-bound FOXP1.
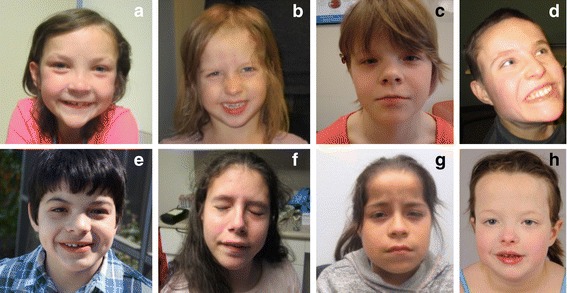
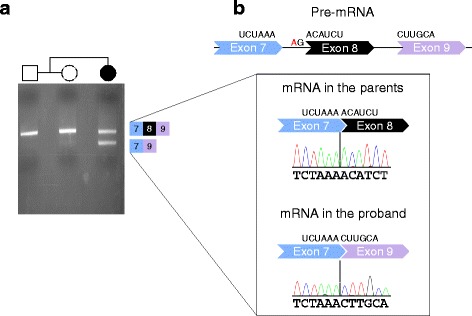
We have identified nine de novo mutations, including three frameshift, one nonsense, one mutation in an essential splice site resulting in frameshift and insertion of a premature stop codon, three missense, and one in-frame deletion. Reviewing prior literature, we found seven instances of recurrent mutations and another 34 private mutations. The majority of pathogenic missense and in-frame mutations, including all four missense mutations in our cohort, lie in the DNA-binding domain. Through structural analyses, we show that the mutations perturb amino acids necessary for binding to the DNA or interfere with the domain swapping that mediates FOXP1 dimerization. Individuals with FOXP1 syndrome presented with delays in early motor and language milestones, language impairment (expressive language > receptive language), ASD symptoms, visual-motor integration deficits, and complex psychiatric presentations characterized by anxiety, obsessive-compulsive traits, attention deficits, and externalizing symptoms. Medical features included non-specific structural brain abnormalities and dysmorphic features, endocrine and gastrointestinal problems, sleep disturbances, and sinopulmonary infections.
This study identifies novel mutations associated with FOXP1 syndrome, identifies recurrent mutations, and demonstrates significant clustering of missense mutations in the DNA-binding domain. Clinical findings confirm the role plays in development across multiple domains of functioning. The genetic findings can be incorporated into clinical genetics practice to improve accurate genetic diagnosis of FOXP1 syndrome and the clinical findings can inform monitoring and treatment of individuals with FOXP1 syndrome.
叉头框蛋白P1(FOXP1)基因的单倍剂量不足会导致一种名为FOXP1综合征的神经发育障碍。先前对携带FOXP1突变和缺失的个体进行的研究已经描述了自闭症谱系障碍(ASD)特征、智力残疾、语言障碍和精神症状的存在。本研究的目的是全面描述FOXP1综合征的遗传和临床谱系。这是第一项使用一系列标准化临床评估对多名FOXP1综合征患者的基因型-表型关系进行前瞻性研究。
获取并分析了9名年龄在5至17岁之间携带FOXP1突变的儿童和青少年的遗传和临床数据。表型特征包括金标准ASD测试以及认知、适应性行为、语言、运动和视动整合技能的常模参照测量。此外,一个多学科临床医生团队完成了精神、医学、神经和畸形学检查。还对已报道的病例进行了全面回顾。所有错义突变和框内突变都映射到与DNA结合的FOXP1的三维结构上。
我们鉴定出9种新发突变,包括3种移码突变、1种无义突变、1种位于关键剪接位点导致移码并插入过早终止密码子的突变、3种错义突变和1种框内缺失。回顾先前的文献,我们发现了7例复发性突变和另外34种私人突变。大多数致病性错义突变和框内突变,包括我们队列中的所有4种错义突变,都位于DNA结合域。通过结构分析,我们表明这些突变扰乱了与DNA结合所需的氨基酸,或干扰了介导FOXP1二聚化的结构域交换。FOXP1综合征患者表现出早期运动和语言发育里程碑延迟、语言障碍(表达性语言>接受性语言)、ASD症状、视动整合缺陷以及以焦虑、强迫特征、注意力缺陷和外化症状为特征的复杂精神症状。医学特征包括非特异性脑结构异常和畸形特征、内分泌和胃肠道问题、睡眠障碍以及鼻窦肺部感染。
本研究鉴定出与FOXP1综合征相关的新的FOXP1突变,确定了复发性突变,并证明错义突变在DNA结合域中显著聚集。临床发现证实了FOXP1在多个功能领域发育中的作用。这些遗传发现可纳入临床遗传学实践,以改善FOXP1综合征的准确基因诊断,临床发现可为FOXP1综合征患者的监测和治疗提供参考。