Department of Medicine, University of Miami Miller School of Medicine, FL, USA.
Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, FL, USA.
Mol Oncol. 2017 Dec;11(12):1806-1825. doi: 10.1002/1878-0261.12151. Epub 2017 Nov 16.
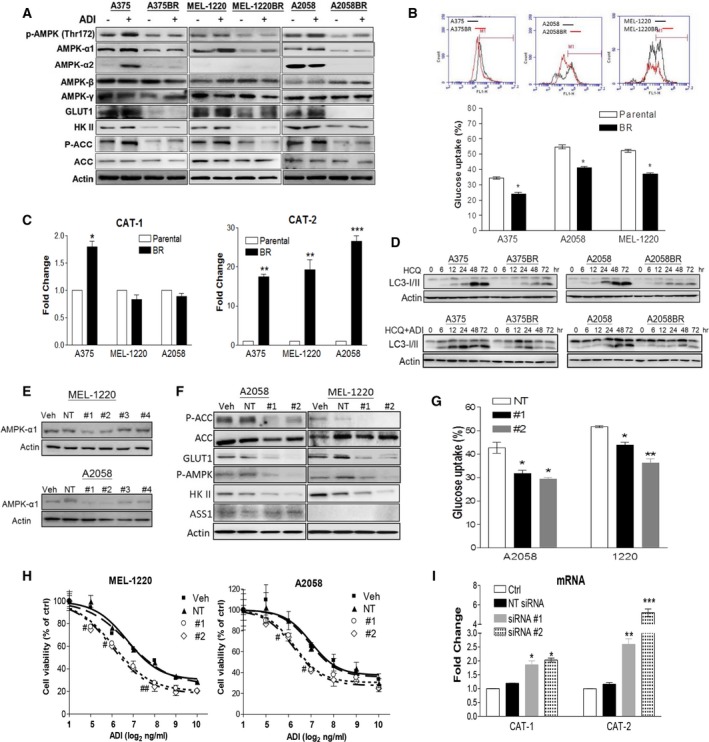
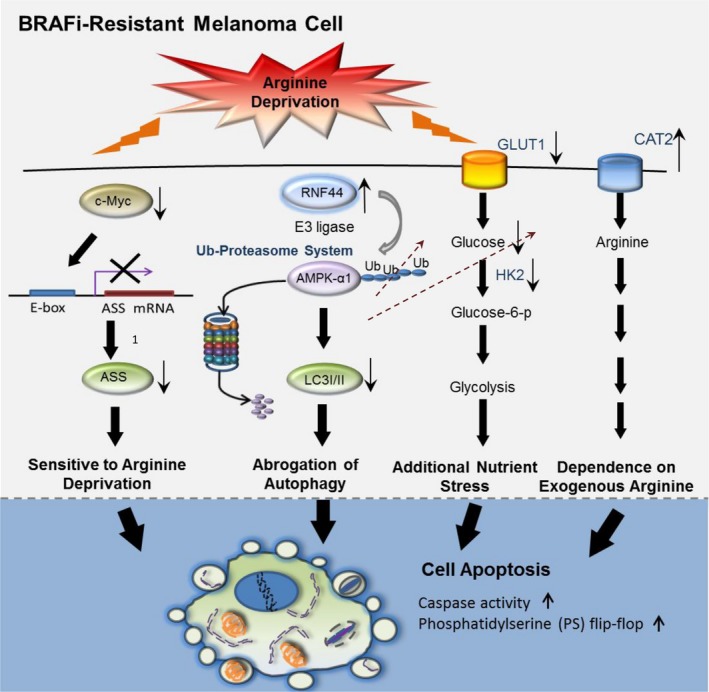
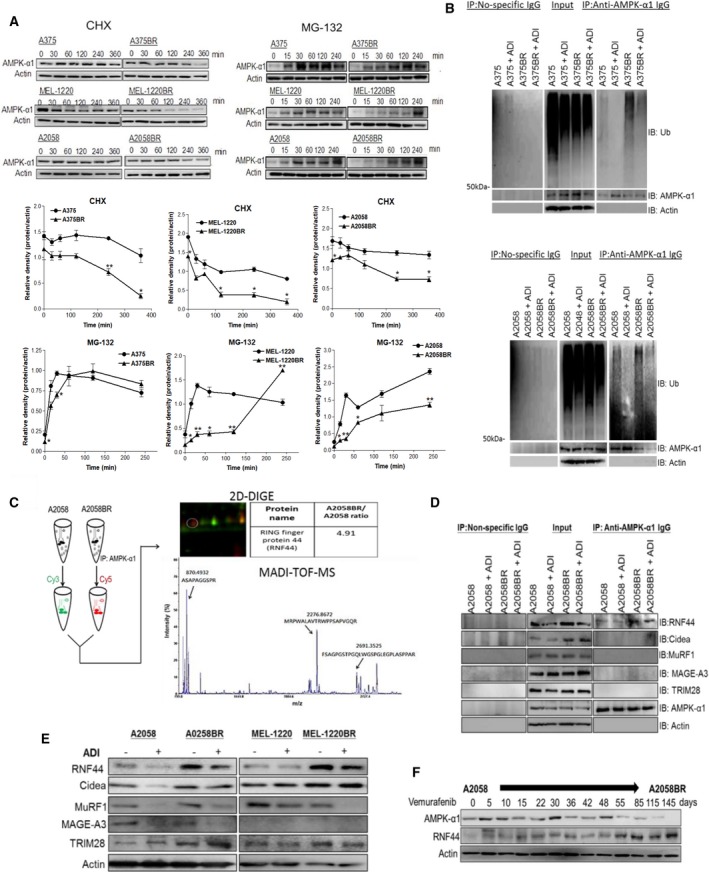
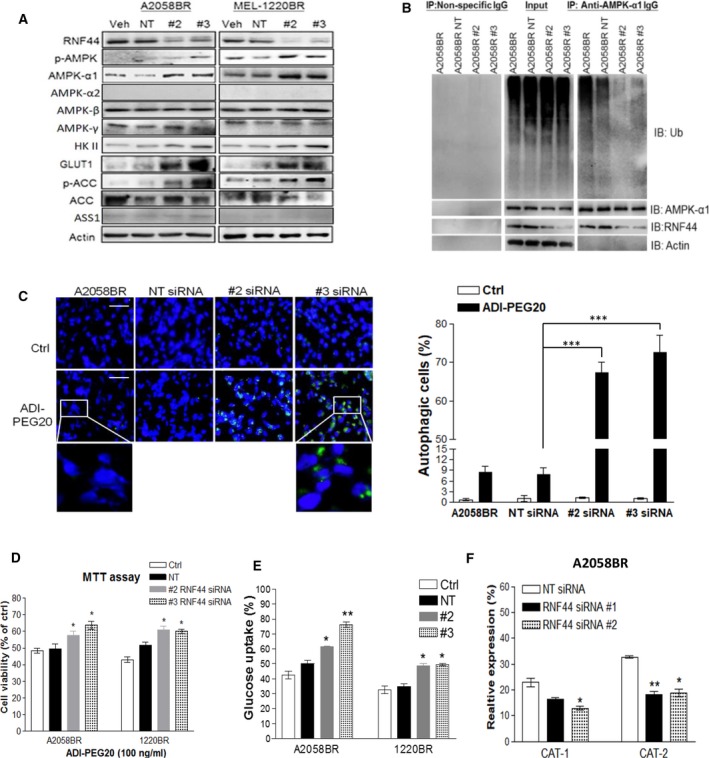
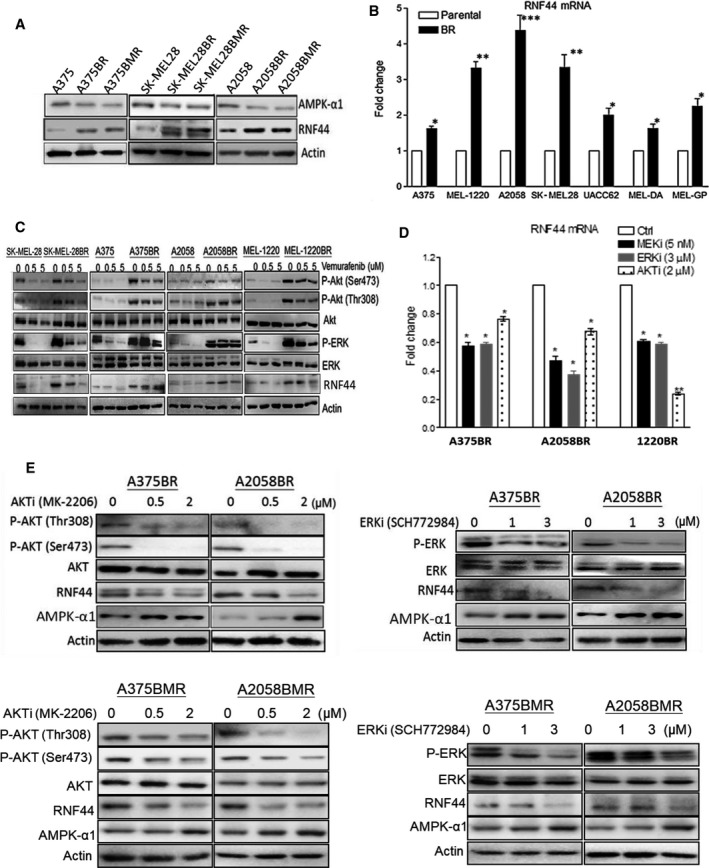
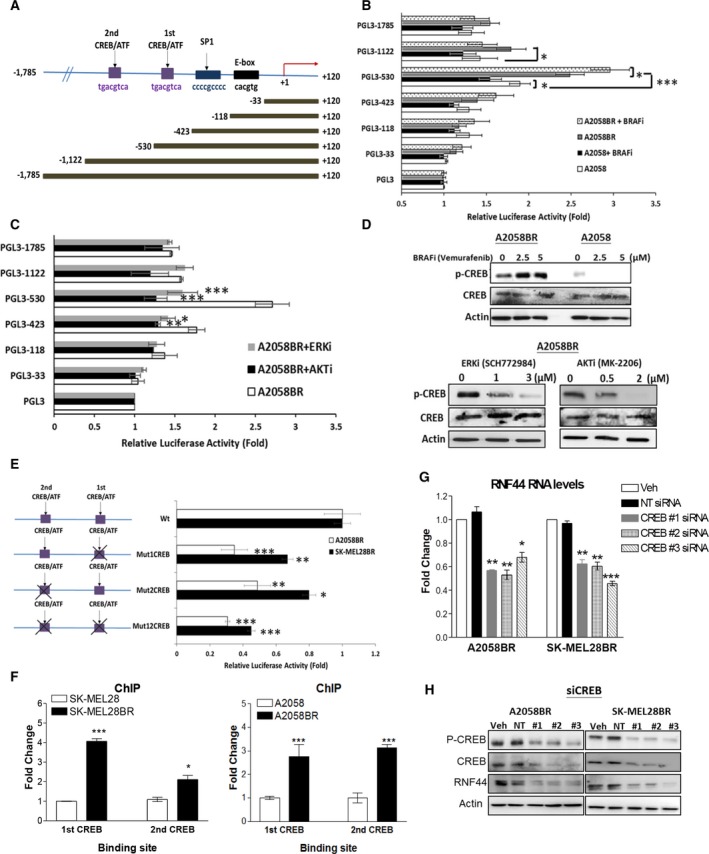
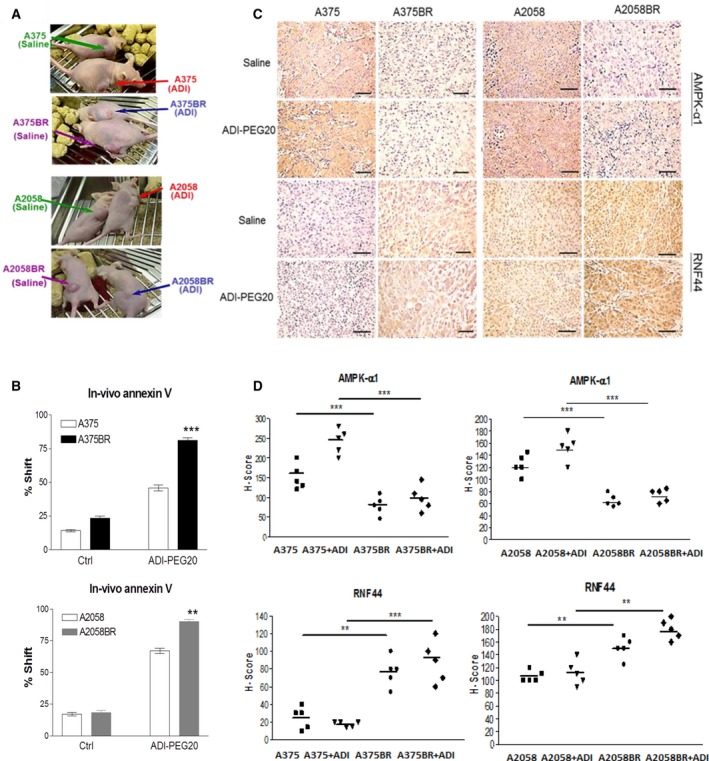
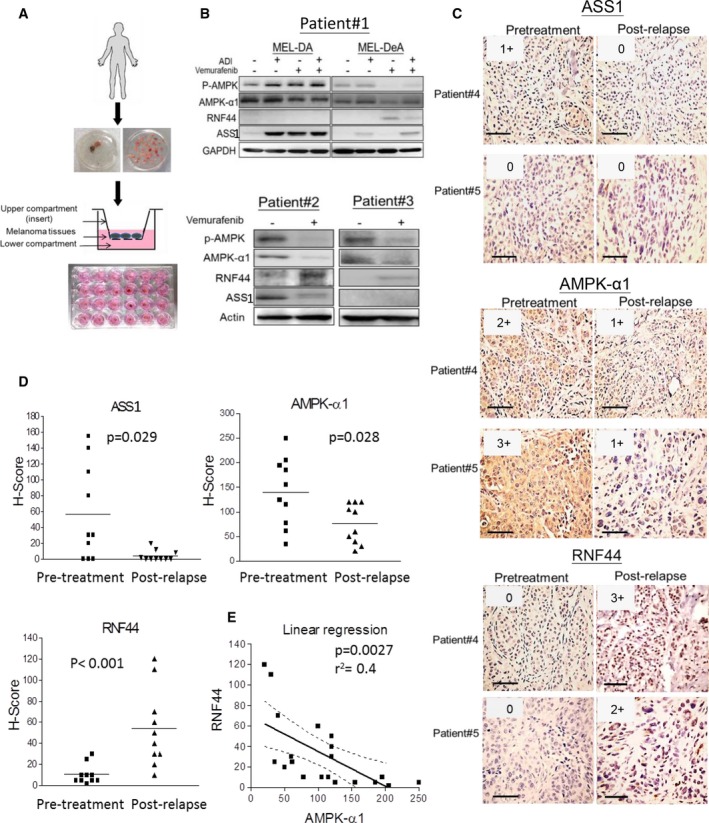
Melanomas harboring BRAF mutation (V600E) are known to recur frequently following treatment with BRAF inhibitors (BRAFi) despite a high initial response rate. Our previous study has uncovered that BRAFi-resistant melanoma (BR) cells are vulnerable to arginine deprivation. It has been reported that naïve melanoma cells undergo autophagy and re-express argininosuccinate synthetase 1 (ASS1) to enable them to synthesize arginine for survival when encountering arginine deprivation. Abolishing these two factors in BR cells confers sensitivity to arginine deprivation. In this report, we further demonstrated that downregulation of AMPK-α1 in BR cells is a major factor contributing to impairment of autophagy as evidenced by decreased autophagosome formation. These BR cells also showed a metabolic shift from glucose to arginine dependence, which was supported by decreased expressions of GLUT1 (glucose transporter) and hexokinase II (HKII) coupled with less glucose uptake but high levels of arginine transporter CAT-2 expression. Furthermore, silencing CAT-2 expression also distinctly attenuated BR cell proliferation. Notably, when naïve melanoma cells became BR cells by long-term exposure to BRAFi, a stepwise degradation of AMPK-α1 was initiated via ubiquitin-proteasome system (UPS). We discovered that a novel E3 ligase, RING finger 44 (RNF44), is responsible for promoting AMPK-α1 degradation in BR cells. RNF44 expression in BR cells was upregulated by transcription factor CREB triggered by hyperactivation of ERK/AKT. High levels of RNF44 corresponding to low levels of AMPK-α1 appeared in BR xenografts and melanoma tumor samples from BR and BRAFi/MEK inhibitor (MEKi)-resistant (BMR) melanoma patients. Similar to BR cells, BMR cells were also sensitive to arginine deprivation. Our study provides a novel insight into the mechanism whereby BRAFi or BRAFi/MEKi resistance drives proteasomal degradation of AMPK-α1 and consequently regulates autophagy and metabolic reprogramming in melanoma cells.
黑色素瘤中存在 BRAF 突变(V600E),尽管初始反应率很高,但在用 BRAF 抑制剂(BRAFi)治疗后仍会频繁复发。我们之前的研究发现,BRAFi 耐药性黑色素瘤(BR)细胞对精氨酸缺乏敏感。据报道,原始黑色素瘤细胞会发生自噬并重新表达精氨酸合成酶 1(ASS1),以使它们在遇到精氨酸缺乏时能够合成精氨酸以存活。在 BR 细胞中消除这两个因素可使细胞对精氨酸缺乏敏感。在本报告中,我们进一步证明 BR 细胞中 AMPK-α1 的下调是自噬受损的主要因素,这表现在自噬体形成减少。这些 BR 细胞还表现出从葡萄糖依赖到精氨酸依赖的代谢转变,这与葡萄糖转运蛋白 1(GLUT1)和己糖激酶 II(HKII)的表达减少以及葡萄糖摄取减少但精氨酸转运蛋白 CAT-2 表达水平升高有关。此外,沉默 CAT-2 表达也明显抑制了 BR 细胞的增殖。值得注意的是,当原始黑色素瘤细胞通过长期暴露于 BRAFi 成为 BR 细胞时,通过泛素蛋白酶体系统(UPS)开始逐步降解 AMPK-α1。我们发现一种新型 E3 连接酶,环指蛋白 44(RNF44),负责促进 BR 细胞中 AMPK-α1 的降解。BR 细胞中 RNF44 的表达受 ERK/AKT 过度激活触发的转录因子 CREB 上调。在 BR 异种移植物和来自 BR 和 BRAFi/MEKi 耐药(BMR)黑色素瘤患者的黑色素瘤肿瘤样本中,高 RNF44 水平对应于低 AMPK-α1 水平。与 BR 细胞类似,BMR 细胞也对精氨酸缺乏敏感。我们的研究为 BRAFi 或 BRAFi/MEKi 耐药性驱动黑色素瘤细胞中 AMPK-α1 的蛋白酶体降解并由此调节自噬和代谢重编程的机制提供了新的见解。