Bioinformatics Division, Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, 3052, Australia.
Department of Medical Biology, University of Melbourne, Parkville, Victoria, 3010, Australia.
Genome Res. 2017 Dec;27(12):2050-2060. doi: 10.1101/gr.222109.117. Epub 2017 Nov 2.
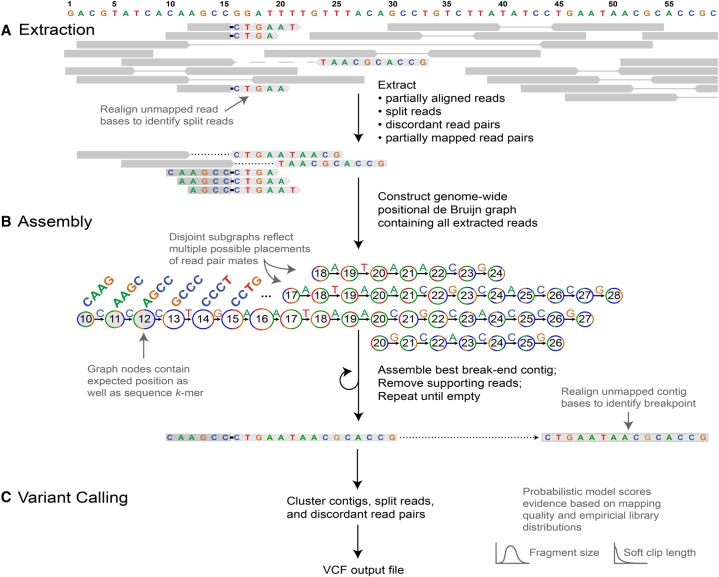
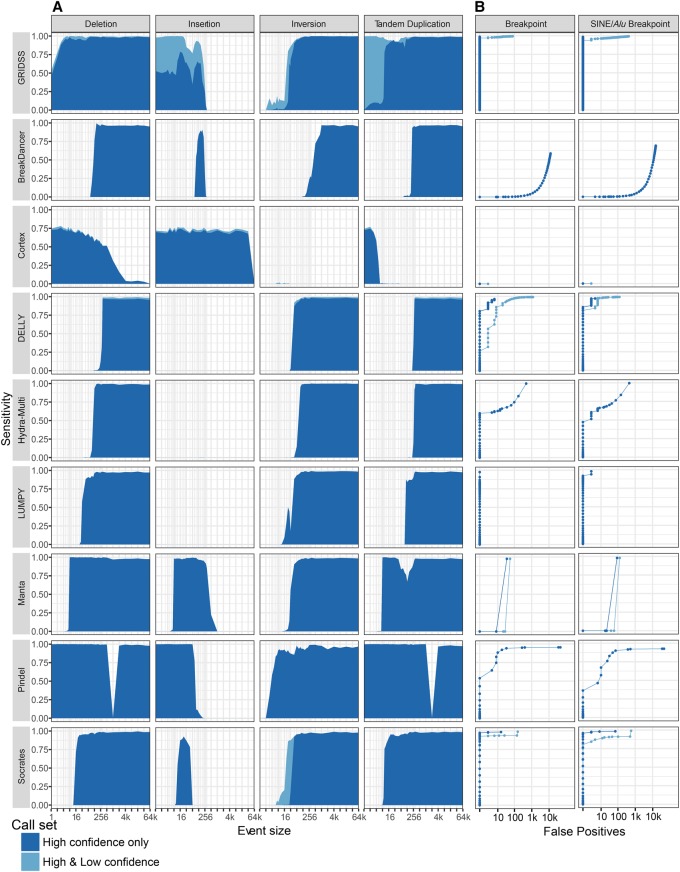
The identification of genomic rearrangements with high sensitivity and specificity using massively parallel sequencing remains a major challenge, particularly in precision medicine and cancer research. Here, we describe a new method for detecting rearrangements, GRIDSS (Genome Rearrangement IDentification Software Suite). GRIDSS is a multithreaded structural variant (SV) caller that performs efficient genome-wide break-end assembly prior to variant calling using a novel positional de Bruijn graph-based assembler. By combining assembly, split read, and read pair evidence using a probabilistic scoring, GRIDSS achieves high sensitivity and specificity on simulated, cell line, and patient tumor data, recently winning SV subchallenge #5 of the ICGC-TCGA DREAM8.5 Somatic Mutation Calling Challenge. On human cell line data, GRIDSS halves the false discovery rate compared to other recent methods while matching or exceeding their sensitivity. GRIDSS identifies nontemplate sequence insertions, microhomologies, and large imperfect homologies, estimates a quality score for each breakpoint, stratifies calls into high or low confidence, and supports multisample analysis.
使用大规模平行测序技术高灵敏度和特异性地识别基因组重排仍然是一个主要挑战,特别是在精准医学和癌症研究中。在这里,我们描述了一种新的重排检测方法,即 GRIDSS(基因组重排识别软件套件)。GRIDSS 是一种多线程结构变体(SV)调用器,在使用新型基于位置的 de Bruijn 图的组装器进行变体调用之前,高效地进行全基因组断点端组装。通过使用概率评分结合组装、分裂读取和读取对证据,GRIDSS 在模拟、细胞系和患者肿瘤数据上实现了高灵敏度和特异性,最近在 ICGC-TCGA DREAM8.5 体细胞突变调用挑战的 SV 子挑战 #5 中获胜。在人类细胞系数据上,与其他最近的方法相比,GRIDSS 将假阳性率降低了一半,同时匹配或超过了它们的灵敏度。GRIDSS 可以识别非模板序列插入、微同源性和大的不完全同源性,为每个断点估计一个质量得分,将调用分为高置信度或低置信度,并支持多样本分析。