Liu Jun, Bhadra Malini, Sinnakannu Joanna Rajeswary, Yue Wan Lin, Tan Cheryl Weiqi, Rigo Frank, Ong S Tiong, Roca Xavier
School of Biological Sciences, Nanyang Technological University, Singapore.
Cancer and Stem Cell Biology Signature Research Programme, Duke-NUS Medical School, Singapore.
Oncotarget. 2017 Sep 6;8(44):77567-77585. doi: 10.18632/oncotarget.20658. eCollection 2017 Sep 29.
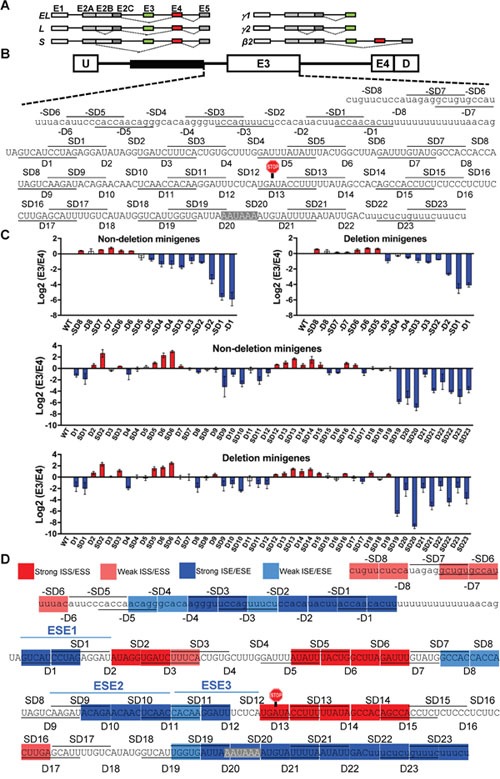
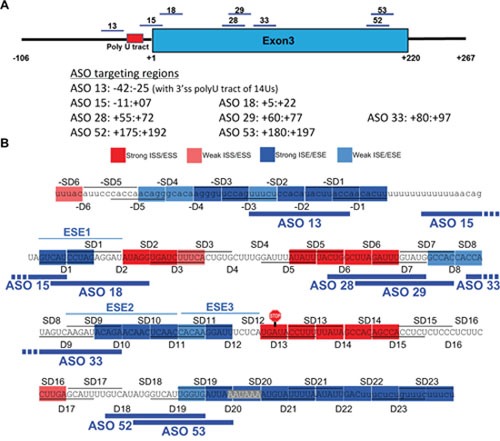
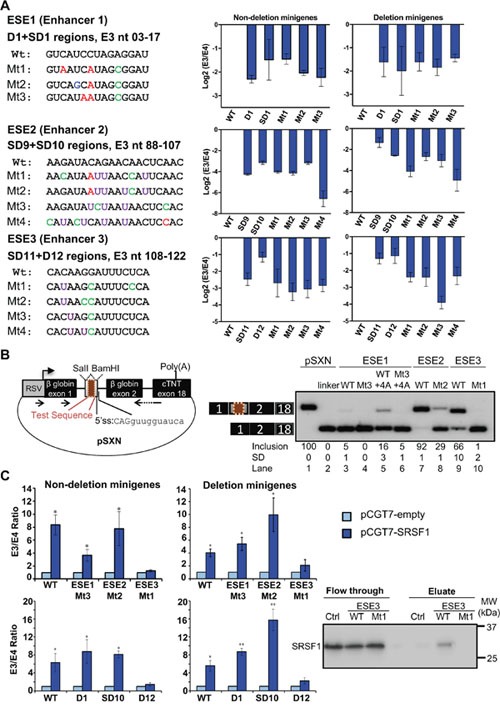
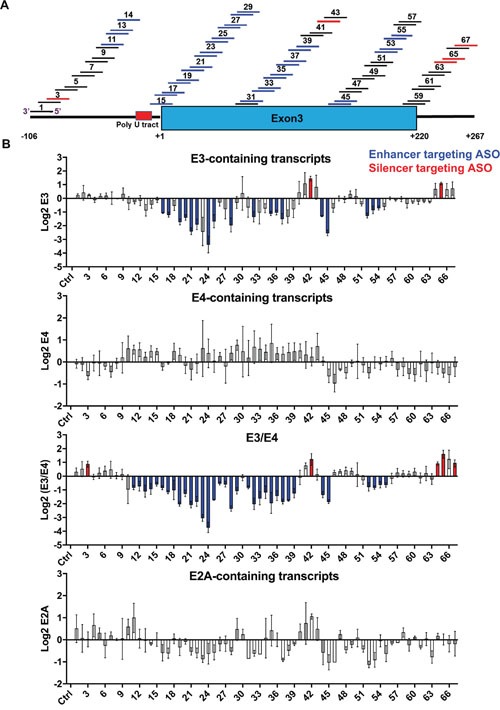
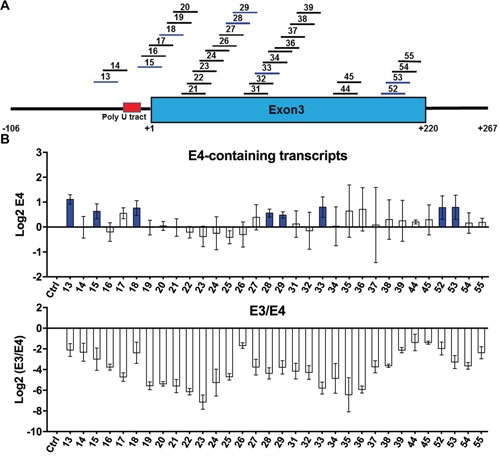
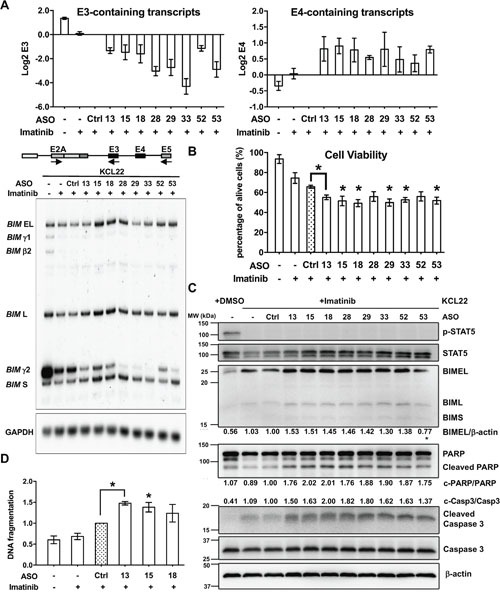
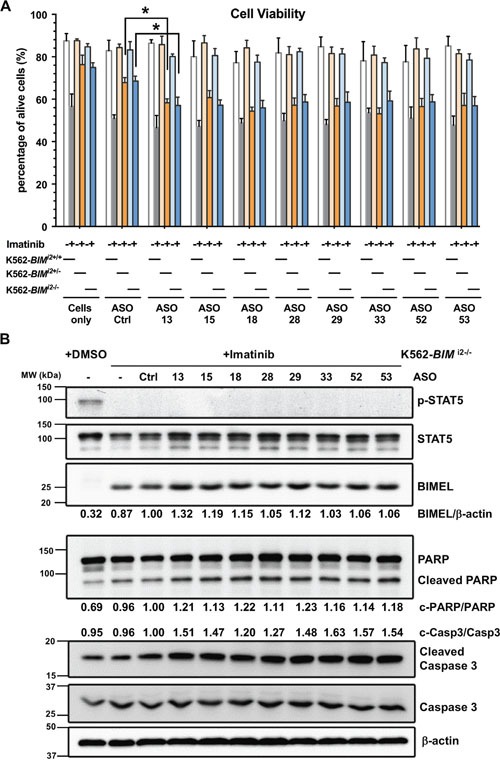
Many tyrosine kinase-driven cancers, including chronic myeloid leukemia (CML), are characterized by high response rates to specific tyrosine kinase inhibitors (TKIs) like imatinib. In East Asians, primary imatinib resistance is caused by a deletion polymorphism in Intron 2 of the gene, whose product is required for TKI-induced apoptosis. The deletion biases splicing from exon 4 to exon 3, generating splice isoforms lacking the exon 4-encoded pro-apoptotic BH3 domain, which impairs the ability of TKIs to induce apoptosis. We sought to identify splice-switching antisense oligonucleotides (ASOs) that block exon 3 but enhance exon 4 splicing, and thereby resensitize deletion-containing cancers to imatinib. First, we mapped multiple -acting splicing elements around exon 3 by minigene mutations, and found an exonic splicing enhancer acting via SRSF1. Second, by a systematic ASO walk, we isolated ASOs that corrected the aberrant splicing. Eight of 67 ASOs increased exon 4 levels in deletion-containing cells, and restored imatinib-induced apoptosis and TKI sensitivity. This proof-of-principle study proves that resistant CML cells by deletion polymorphism can be resensitized to imatinib via splice-switching ASOs. Future optimizations might yield a therapeutic ASO as precision-medicine adjuvant treatment for -polymorphism-associated TKI-resistant CML and other cancers.
许多酪氨酸激酶驱动的癌症,包括慢性粒细胞白血病(CML),其特征是对伊马替尼等特定酪氨酸激酶抑制剂(TKIs)有较高的反应率。在东亚人中,原发性伊马替尼耐药是由该基因第2内含子的缺失多态性引起的,其产物是TKI诱导凋亡所必需的。这种缺失偏向于外显子4到外显子3的剪接,产生缺乏外显子4编码的促凋亡BH3结构域的剪接异构体,这损害了TKIs诱导凋亡的能力。我们试图鉴定能够阻断外显子3但增强外显子4剪接的剪接转换反义寡核苷酸(ASOs),从而使含缺失的癌症对伊马替尼重新敏感。首先,我们通过小基因突变绘制了外显子3周围的多个剪接作用元件,并发现了一个通过SRSF1起作用的外显子剪接增强子。其次,通过系统的ASO筛选,我们分离出了能够纠正异常剪接的ASOs。67个ASOs中有8个提高了含缺失细胞中外显子4的水平,并恢复了伊马替尼诱导的凋亡和TKI敏感性。这项原理验证研究证明,通过缺失多态性产生耐药的CML细胞可以通过剪接转换ASOs对伊马替尼重新敏感。未来的优化可能会产生一种治疗性ASO,作为针对多态性相关TKI耐药CML和其他癌症的精准医学辅助治疗。