School of Computer Science and Technology, Tianjin University, Tianjin 300350, China.
School of Chemical Engineering, Tianjin University, Tianjin 300350, China.
Int J Mol Sci. 2017 Nov 14;18(11):2400. doi: 10.3390/ijms18112400.
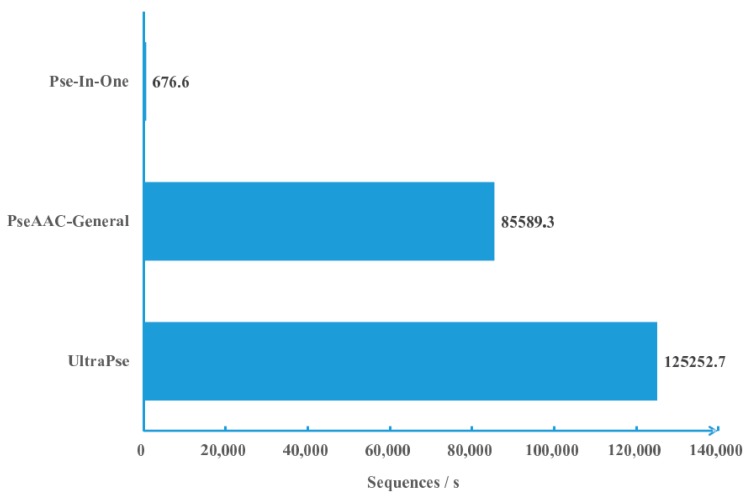
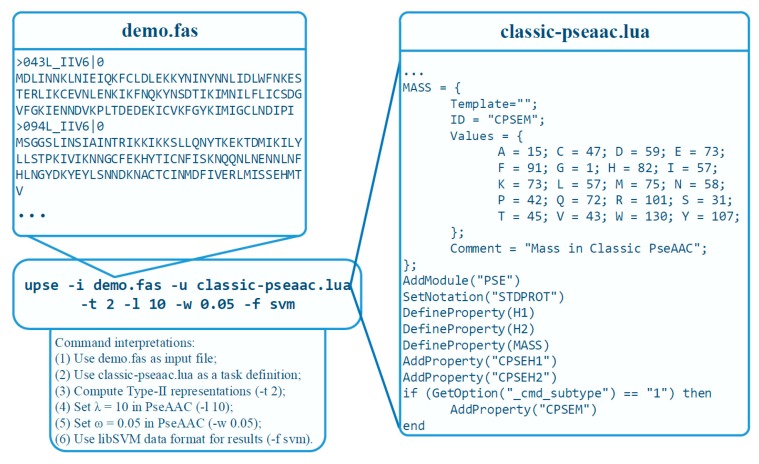
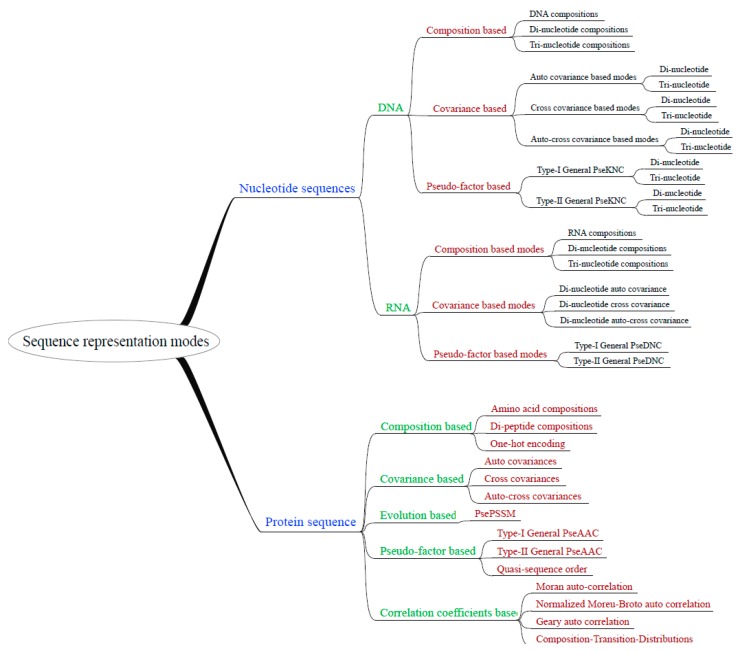
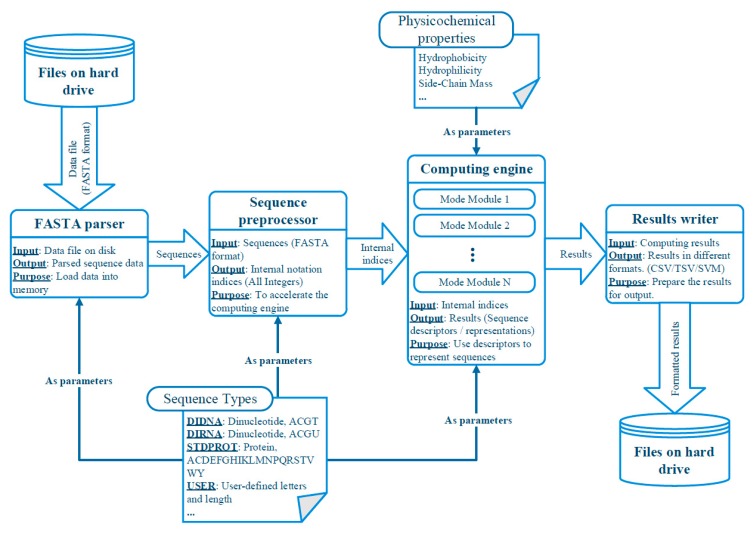
With the avalanche of biological sequences in public databases, one of the most challenging problems in computational biology is to predict their biological functions and cellular attributes. Most of the existing prediction algorithms can only handle fixed-length numerical vectors. Therefore, it is important to be able to represent biological sequences with various lengths using fixed-length numerical vectors. Although several algorithms, as well as software implementations, have been developed to address this problem, these existing programs can only provide a fixed number of representation modes. Every time a new sequence representation mode is developed, a new program will be needed. In this paper, we propose the UltraPse as a universal software platform for this problem. The function of the UltraPse is not only to generate various existing sequence representation modes, but also to simplify all future programming works in developing novel representation modes. The extensibility of UltraPse is particularly enhanced. It allows the users to define their own representation mode, their own physicochemical properties, or even their own types of biological sequences. Moreover, UltraPse is also the fastest software of its kind. The source code package, as well as the executables for both Linux and Windows platforms, can be downloaded from the GitHub repository.
随着公共数据库中生物序列的大量涌现,计算生物学中最具挑战性的问题之一是预测它们的生物学功能和细胞属性。大多数现有的预测算法只能处理固定长度的数值向量。因此,能够使用固定长度的数值向量表示各种长度的生物序列非常重要。尽管已经开发了几种算法和软件实现来解决这个问题,但这些现有的程序只能提供固定数量的表示模式。每次开发新的序列表示模式时,都需要一个新的程序。在本文中,我们提出了 UltraPse 作为解决这个问题的通用软件平台。UltraPse 的功能不仅是生成各种现有的序列表示模式,还简化了开发新表示模式的所有未来编程工作。UltraPse 的可扩展性得到了特别增强。它允许用户定义自己的表示模式、自己的物理化学性质,甚至自己类型的生物序列。此外,UltraPse 也是同类软件中最快的。源代码包以及适用于 Linux 和 Windows 平台的可执行文件均可从 GitHub 存储库下载。